
脑腱黄瘤病2例报告并文献复习
2023-06-06 08:54余婷杜星宇刘海军
中国神经免疫学和神经病学杂志 2023年3期
余婷 杜星宇 刘海军
脑腱黄瘤病(cerebrotendinous xanthomatosis,CTX)是一种由胆固醇27-羟化酶(sterol 27-hydroxylase,CYP27A)基因突变引起的常染色体隐性遗传性脂质贮积症[1],其全球发病率估计在(3~5)/10万,主要临床表现包括婴幼儿腹泻、青少年白内障、肌腱黄色瘤以及神经功能障碍等[2]。该病较为罕见且发病隐匿,症状多样,特别是在疾病早期,其症状往往为非特异性,容易导致误诊、漏诊。研究发现,CTX患者确诊时的平均年龄为35岁,延迟诊断可达16年[3]。现报道作者医院收治的2例CTX患者并结合文献进行复习,总结CTX的发病机制、临床表现、辅助检查及治疗情况,为进一步认识该病提供参考。
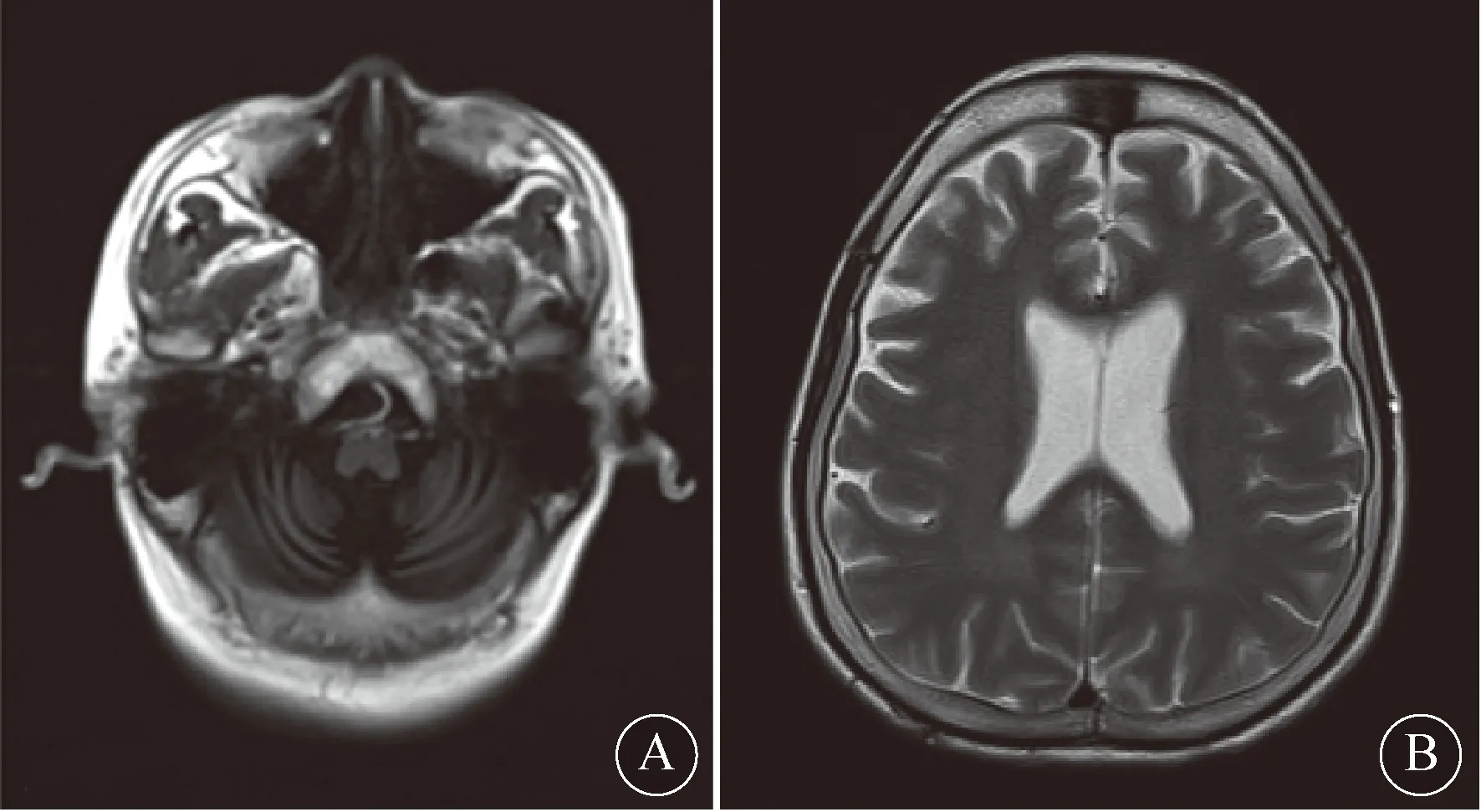
1 病例报告病例1:患者女,41岁,初中学历,成绩中等。因“双侧足后跟包块渐进性增大18年,伴步态不稳2个月”于2018-10-30入院。18年前发现双侧足后跟部1 cm×2 cm包块,局部无压痛,质软,可活动,未见明显疼痛、红肿,包块逐渐增大,大小约为15 cm×6 cm×4 cm,质稍硬,有压痛,固定,能与骨组织鉴别。近2个月来出现步态不稳,易跌倒,未见明显精神异常。无头晕、头痛、肢体麻木、饮水呛咳、意识障碍、听力下降等。患者自幼出现视力减退,逐渐加重,于当地医院诊断为“先天性白内障”,曾行2次手术治疗,目前双眼视力明显下降,以左侧为重。20余年前有“脑膜炎”病史,现遗留智力下降。入院查体:生命体征平稳,意识清楚,查体合作。粗测视力、听力减退,双瞳孔等大等圆,对光反射稍迟钝。四肢肌力正常,双下肢小腿中部至足跟部可见15 cm×6 cm×4 cm包块组织,感觉对称存在。四肢腱反射亢进,脑膜刺激征(-),Romberg征(+),跟膝胫试验阳性,右手Hoffman征(+),双侧Babinski征(+)。血生化检查:总胆汁酸16.78 μmol/L,白蛋白37.4 g/L;A/G 1.17,前白蛋白180 mg/L,抗核抗体1∶100(弱阳性),总胆固醇、低密度脂蛋白、高密度脂蛋白、同型半胱氨酸等均在正常范围。血、尿、便常规以及肾功能、血电解质检查结果均无明显异常。头颅CT检查:左侧小脑低密度病变。踝关节MRI检查:右足轻度骨质增生,左足未见异常。头颅MRI检查:小脑萎缩并软化灶和变性灶(图1)。脑MRS检查提示小脑病变胶质增生和神经元破坏。行左足跟部软组织切除并行活检病理检查,结果提示:增生玻变结缔组织,局部见胆固醇结晶及多核巨细胞反应(图2)。结合临床表现及辅助检查,考虑CTX可能。建议患者行基因CYP27A1检测,但患者拒绝检测。住院期间给予B族维生素营养神经、复方脑肽节苷脂改善脑功能代谢等对症支持治疗,患者步态不稳症状好转。2018-11-11患者出院,给予牛磺熊去氧胆酸口服对症降胆酸治疗。出院1个月后电话随诊,患者症状较前好转。2022-3-6复查颅脑MRI提示:小脑萎缩及多发异常信号,符合脑腱黄瘤病表现。

注:A:T1像;B:T2像
病例2:患者男,32岁。因“步态不稳2年余,双下肢无力10 d”于2019-11-29入院。2年前无明显诱因出现步态不稳,拄物可行走,行走数分钟即感疲劳。曾就诊当地医院行颅脑MRI检查,结果显示双侧额叶及侧脑室后角旁白质缺血灶;右踝关节MRI检查显示右侧跟腱病变。考虑CTX可能。10 d前上述症状加重伴双下肢无力,症状逐渐进展并累及双上肢。无头晕、头痛、肢体麻木、肌肉疼痛、饮水呛咳、意识障碍等。幼时有“脑膜炎”病史,治疗后遗有精神发育迟缓。自幼视力差,于当地医院诊断为“白内障”,未行正规治疗,现遗留双眼视力下降。2年前因胆囊结石行“胆囊切除术”。余既往史无特殊。入院查体:生命体征平稳,体形消瘦。意识清楚,计算力、记忆力下降,反应迟钝,吐词不清。四肢肌肉无萎缩,右侧小腿下方可见长条状隆起性包块,质硬,活动度差,有触痛。四肢肌力2级,肌张力减弱,脑膜刺激征(-)。四肢感觉检查无法配合。四肢腱反射减弱,Romberg征(+)。血常规检查:白细胞总数21.47×109/L,红细胞总数4.29×1012/L,血小板总数629×109/L;血生化检查:乳酸脱氢酶449 U/L,α-羟丁酸脱氢酶274 U/L,肌红蛋白187.20 ng/mL,总胆汁酸49.51 μmol/L,总胆红素48.3 μmol/L,直接胆红素31.6 μmol/L。腰穿脑脊液检查:颅内压150 mmH2O(1 mmH2O=9.8 Pa),氯127.0 mmol/L,葡萄糖3.99 mmol/L,乳酸脱氢酶38 U/L,蛋白1 101 mg/L,总细胞计数225×106/L,其余脑脊液检查未见异常。头颅MRI检查:右侧额叶、左侧枕叶脑白质少许缺血灶,脑萎缩征象(图3)。外送副肿瘤综合征抗体、自身免疫周围神经病抗体、血清及脑脊液寡克隆分析结果均为阴性。外送CYP27A1基因检测:CYP27A1基因2q35区带2个杂合变异,其中2号内含子c.379C>T为精氨酸错义突变,6号内含子c.1184G>A为精氨酸错义突变。根据Gallu等总结的数据库显示,c.379C>T和c.1184G>A均为CTX的潜在致病基因。患者携带CYP27A1基因的两个杂合致病变异,需结合临床综合判断。患者拒绝行病灶组织病理活检。半个月后行腰穿复查脑脊液:颅内压160 mmH2O,蛋白3 036 mg/L。诊断:CTX。给予胆汁酸替代治疗后好转出院。患者出院后未再随诊。
注:A:T1像;B:T2像
2 讨论CTX是一种罕见的常染色体隐性遗传病,由CYP27A1基因变异引起。CYP7A1是胆汁酸合成经典途径的关键酶,其功能丧失可导致胆汁酸、鹅去氧胆酸(chenodeoxycholic acid)和胆酸(cholic acid,CA)的缺乏,同时亦可导致胆汁酸中间体7α-羟基4-胆甾烯-3-酮(7α-hydroxy-4-cholesten-3-one,7αC4)水平升高。CTX患者血液7αC4水平可较健康者高100倍[4]。这些异常的代谢产物在血浆和组织中积聚,特别是大脑(主要是白质)、晶状体和肌腱[2],使患者产生特征性的临床表现,包括神经系统功能障碍、白内障以及肌腱黄色瘤等。
CTX起病隐匿,症状多样,早期症状常不典型。CTX常常累及多个系统,许多患者从婴幼儿时期即有原因不明的腹泻,青少年时出现白内障,并遗留视力障碍。神经系统症状表现成进行性发展,早期可出现认知功能障碍、小脑性共济失调和癫痫,晚期可出现锥体束体征、进行性痉挛性截瘫、小脑共济失调和构音障碍、外周性多发性神经病,以及更罕见的运动障碍等[5]。该组2例患者均有白内障病史,临床表现主要以神经系统为主,表现为步态不稳、共济失调以及认知功能障碍,颅脑MRI提示小脑萎缩,查体可见Hoffmann征及Babinski征阳性。这与既往报道的CTX临床症状相一致[6]。该组患者早期以婴幼儿腹泻、青少年白内障这些非神经系统表现为主。有研究显示临床未能充分认识CTX的这些非神经系统症状是其诊断延迟的主要原因[7]。因此,儿科和眼科医生对CTX的早期诊断具有重要作用。
目前,基因检测是CTX诊断的“金标准”,主要通过检测CYP7A1基因确诊。CYP27A1基因包含9个外显子和8个内含子。Gallu等[8]总结了CYP27A1基因的1~8外显子的多样性,共表达了49个不同类型的突变。该组病例2检测到2个杂合突变,即Ri27W(p.379C>T)及R395H(p.1184G>A),均为数据库[8]中已知的CTX致病位点,患者表现为典型的CTX症状,如白内障、腱黄瘤及神经系统症状等。
CTX的标准治疗是口服鹅去氧胆酸治疗,鹅去氧胆酸可通过负反馈途径抑制胆汁酸合成来防止胆固醇的积累同时抑制CYP7A1的表达[9]。Stelten等[10]研究表明,早期开始治疗可以逆转甚至预防CTX的神经系统症状,提示CTX早期诊断的重要性。而延误诊断和治疗可导致更差的预后、不可逆的神经损伤和严重的残疾[11]。
猜你喜欢
保健医苑(2023年2期)2023-03-15
现代临床医学(2022年4期)2022-09-29
理化检验-化学分册(2021年10期)2021-11-29
肝博士(2020年5期)2021-01-18
中西医结合肝病杂志(2020年2期)2020-10-27
电子制作(2018年18期)2018-11-14
自动化学报(2018年6期)2018-07-23
中国卫生标准管理(2015年16期)2016-01-20
中国当代医药(2015年9期)2015-03-01
发明与创新(2015年33期)2015-02-27
