
仅表现声带麻痹的MuSK抗体阳性重症肌无力一例并文献复习
2023-06-06 08:54:56杨梦婷姜梦迪赵凌云殷剑侯世芳张华
中国神经免疫学和神经病学杂志 2023年3期
杨梦婷 姜梦迪 赵凌云 殷剑 侯世芳 张华
重症肌无力(myasthenia gravis,MG)是累及神经肌肉接头的获得性自身免疫疾病,临床表现为局部或全身特定骨骼肌的病态易疲劳和波动性肌无力[1]。其发病多由骨骼肌乙酰胆碱受体(AChR)抗体引起,其他非AChR成分如肌肉特异性酪氨酸激酶(muscle skeletal receptor tyrosine kinase,MuSK)也可作为免疫攻击的目标引起MG[2]。抗MuSK抗体阳性MG多为女性,多于30岁以后起病,表现为选择性咽喉肌、颈部肌肉、呼吸肌无力,易出现肌无力危象,对抗胆碱酯酶药物的敏感性差[2-3]。单独累及声带的MG患者极为罕见,多由于喉内肌的神经肌肉接头传递障碍引起声带活动障碍,出现双侧声带麻痹导致上气道阻塞,甚至危及生命[4-5]。抗MuSK抗体阳性的MG患者延髓部肌肉受累多见,目前国外有少数出现声带麻痹的个案报道[6-10]。现报道作者医院收治的1例长期仅表现为双侧声带麻痹的抗MuSK抗体阳性MG患者的临床特点,并结合文献进行复习,以提高对该病的认识。
1 对象和方法
1.1 病例报告患者女,62岁。因“发音不清11年,呼吸困难3年余,加重1个月余”于2022-4-19入院。11年前出现声音嘶哑、发音不清,发音不清无疲劳及晨轻暮重现象,无肢体无力、吞咽困难、饮水呛咳,无眼睑下垂、复视,无肌肉萎缩及肉跳,曾多次就诊外院未明确诊断。2011-12-16曾在外院就诊,行头颅MRI平扫及增强检查:双侧基底节区及侧脑室体旁多发对称性白质异常信号,增强扫描未见强化(图1)。头MRA检查未见异常。腰穿脑脊液检查:常规、生化未见异常,病原学检查未见异常。血及脑脊液寡克隆区带、碱性髓鞘蛋白(myelin basic protein,MBP)抗体、水通道蛋白4(aquaporin 4,AQP4)抗体均阴性。予改善循环治疗后症状无改善。3年多前出现憋气、喘鸣,多次就诊于外院,血气二氧化碳分压波动在85 mmHg左右,喉镜检查提示双侧声带外展受限。建议行气管插管或切开,患者拒绝。1个月前因患者喘憋加重就诊作者医院。患者既往体健,家族史无特殊。入院查体:意识清楚,构音不清,伴喉鸣,无明显疲劳现象。高级皮层功能正常。双眼睑无下垂,眼球活动正常。面纹对称,伸舌居中,舌肌无萎缩、纤颤。软腭活动可,咽反射灵敏。四肢肌力、张力正常,未见萎缩,共济正常。深浅感觉未见异常。腱反射对称正常引出,病理征阴性。上睑及四肢疲劳试验阴性。入院时血气分析显示二氧化碳分压86 mmHg。喉镜检查显示,双侧声带不能外展,声门间隙1~2 mm(图2)。新斯的明试验时声音嘶哑无明显改善。新斯的明注射前后行喉镜检查:声带运动无改变,声门裂未见增宽。针极肌电图检查结果未见异常,面神经重复电刺激检查显示面神经低频递减,jitter延长。肌无力抗体(放射免疫法)检测:抗MuSK抗体阳性(0.545 nmol/L;正常值<0.05 nmol/L),余抗体阴性。颈部MRI检查未见迷走神经走行路径上的病变。甲状腺B超检查结果正常。头颅MRI检查:脑桥、双侧侧脑室旁周围、额顶颞叶多发异常信号(图2),与10年前外院头颅MRI检查结果大致相同。腰穿脑脊液检查:脑脊液常规、生化结果正常。血及脑脊液自身免疫性脑炎抗体、副肿瘤抗体阴性,血及脑脊液AQP4抗体、髓鞘少突胶质细胞糖蛋白(myelin oligodendrocyte glycoprotein,MOG)抗体、神经胶质原纤维酸性蛋白(glial fibrillary acidic protein,GFAP)抗体阴性。胸部、腹部、盆腔CT平扫及乳腺、甲状腺超声排查未见肿瘤征象。诊断:MuSK抗体阳性MG;双侧声带麻痹;脑白质病变性质待定。建议行气管插管但患者拒绝,给予无创呼吸机辅助通气。加用溴吡斯的明治疗后患者出现腹泻、肌肉束颤,遂停用。按体重0.4 g/(kg·d)给予静注人免疫球蛋白,共5 d。患者发音困难改善不明显,继而给予甲泼尼龙片口服,起始剂量8 mg/d,每3 d增加4 mg至24 mg/d维持,患者症状仍无明显改善,遂给予序贯利妥昔单抗500 mg治疗,每周1次,共4次。患者发音困难、饮水呛咳明显好转。行喉镜检查显示双侧声带外展受限较前好转,约 5~6 mm(图2)。出院前脱离无创呼吸机,二氧化碳分压稳定在60 mmHg以下。复查头颅MRI检查:颅内多发异常信号(图1)。鉴于患者缺乏脑血管病危险因素,故排除血管源性白质病变。脑脊液常规、生化正常,病原学检查阴性排除颅内感染。根据寡克隆区带阴性、多种抗体阴性,排除脱髓鞘性病变。患者否认中毒,故排除中毒代谢因素。脑白质营养不良和脑小血管病相关基因检测未见异常,排除遗传性白质病变。结合患者10年间头颅MRI病灶无变化,免疫治疗后病灶无变化,且缺乏锥体束征、认知障碍、步态异常等白质异常的症状,考虑颅内病灶和声带麻痹关联不大。嘱患者定期监测头颅MRI变化情况。因病情稳定,准予出院。出院后继续口服甲泼尼龙片24 mg治疗(1次/d),1个月后每周减4 mg直至减停。患者发音困难、饮水呛咳完全恢复,无胸闷气短,继续随访中。

注:A、B:2011-12-16行头颅MRI T2 Flair像检查示脑桥脱髓鞘(A)、侧脑室脱髓鞘(B);C、D:2022-4-22行头颅MRI T2 Flair像检查示脑桥(C)、侧脑室旁脱髓鞘(D);E、F:患者经治疗后2022-6-14行头颅MRI T2 Flair像检查示脑桥(E)和侧脑室(F)脱髓鞘
1.2 文献复习检索中国知网、万方数据库、维普数据库、Pubmed、Cochrone图书馆、Embase数据库自建库以来至2022年6月公开发表的表现为声带麻痹的MuSK阳性MG文献,采用主题词和自由词结合的方式进行检索。英文文献检索词:“myasthenia gravis”“MuSK antibodies”“vocal cord paralysis”。中文文献检索词:“重症肌无力”“MuSK抗体”“声带麻痹”。文献纳入标准:(1)表现为声带麻痹的MuSK型MG的个案报道;(2)MG的诊断符合2020年发布的《中国重症肌无力诊断和治疗指南》标准[11];(3)排除数据不完整的研究。由两名研究者完成文献筛查和数据提取,数据提取的主要内容包括:第一作者、发表年份、发病年龄、出现声带麻痹年龄、临床症状、体格检查、喉镜检查、新斯的明试验、抗体检测、肌电图检查、药物治疗、有创呼吸支持、转归等。
2 结果
共检索到符合条件的文献5篇[6-10],共报道5例患者,包括本文报道的患者共6例纳入分析。
2.1 临床特点6例患者中男2例,女4例,平均年龄(45.17±12.09)岁,出现声带麻痹的年龄 24~59岁,平均(47.17±12.61)岁。患者均存在发音、吞咽、呼吸困难,其中2例由发音、吞咽困难进展为呼吸困难,3例首发症状表现为喘鸣、呼吸困难。2例伴有眼外肌受累。3例伴有面部、四肢肌肉无力。1例伴有肌肉萎缩。具体结果见表1。
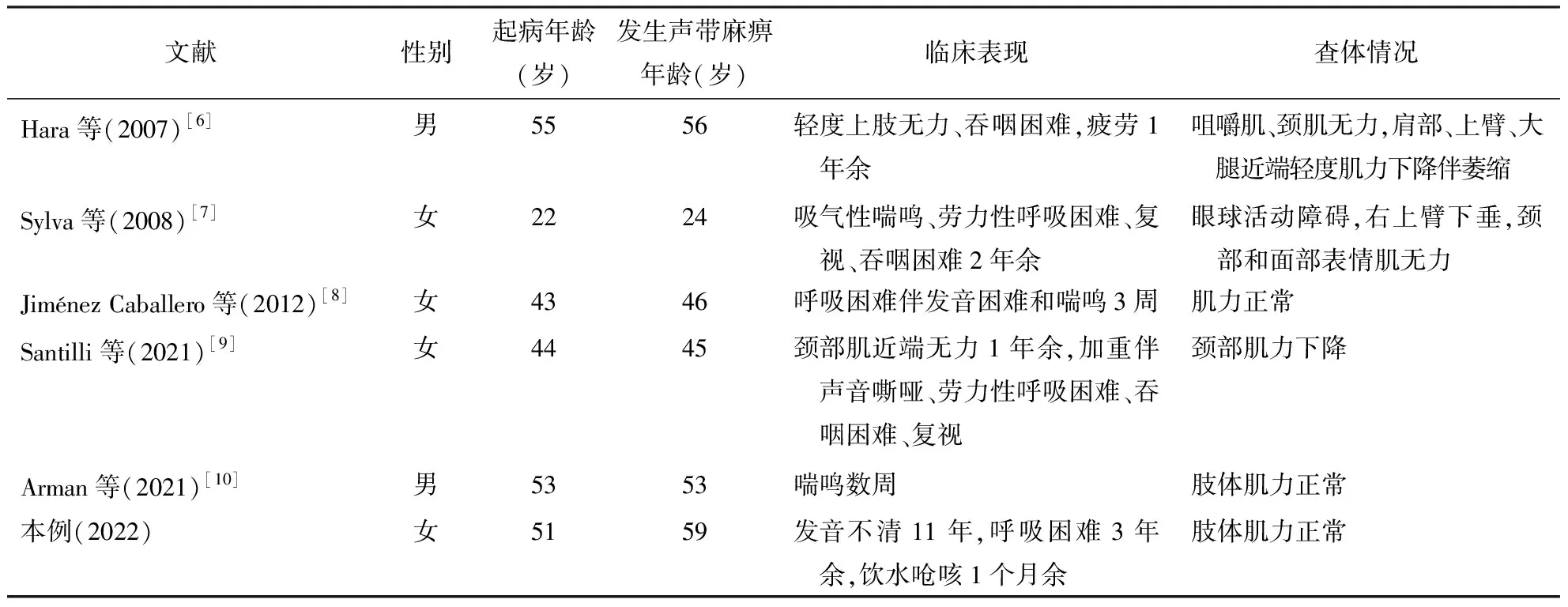
表1 6例表现为双侧声带麻痹的抗MuSK抗体阳性MG患者的临床特点
2.2 辅助检查6例患者行喉镜检查,均表现为声带外展受限。新斯的明试验阳性2例,阴性2例,未说明新斯的明试验情况2例。抗MuSK抗体均阳性,检测方法为放射免疫分析法1例,余5例未说明检测方法。肌电图检查结果显示3例患者仅表现为眼部及面部肌肉低频递减,2例同时有面部及肢体的低频递减,1例未发现异常。2例出现单纤维肌电图的异常,其余患者未给出单纤维肌电图的结果。具体结果见表2。

表2 6例表现为双侧声带麻痹的抗MuSK抗体阳性MG患者辅助检查结果
2.3 治疗及预后4例患者采用溴吡斯的明治疗效果均不佳;2例采用丙种球蛋白治疗,其中1例效果不佳;5例采用皮质类固醇治疗,4例有所改善。2例予以利妥昔单抗治疗有效,3例行气管插管或气管切开有创呼吸支持。经治疗最终6例均有所好转。具体结果见表3。

表3 6例表现为双侧声带麻痹的抗MuSK抗体阳性MG患者辅助检查、治疗及转归
3 讨论
MG是由自身抗体引起的神经肌肉接头疾病,其临床特征为骨骼肌的波动性肌肉无力和易疲劳。70%~80%的MG患者存在针对肌肉烟碱型AChR的自身抗体,近年来在血清AChR抗体阴性的患者中发现了致病抗体MuSK抗体,约占 5%~8%。抗MuSK抗体通过阻断MuSK在神经肌肉接头处诱导AChR聚集和乙酰胆碱酯酶的锚定引起MG症状。MuSK抗体阳性的MG患者临床进展快速,以延髓麻痹起病常见,病程中逐渐累及全身其他部位,肌无力危象发生率高[3,11]。
近年来国外陆续有MuSK抗体阳性MG继发声带麻痹的病例报道。个案分析结果显示,这些患者多表现为声音嘶哑、发音困难,可出现继发于功能性上气道阻塞的喘鸣和呼吸衰竭。声带受累主要影响后环杓肌,导致声带外展无力,引起气流阻力增加。因此,当患者出现伴疲劳现象的发音困难和喘鸣需要警惕MG累及声带的可能性,应及时完善喉镜检查,及时发现患者是否存在声带萎缩和双侧外展受损情况[5]。该例患者存在发音困难而缺乏疲劳现象,提示缺乏疲劳现象并不能排除MG的可能,应引起临床重视。
通过文献复习发现,2例患者对新斯的明试验反应欠佳。临床上MuSK抗体阳性MG对胆碱酯酶类药物反应常不敏感,且与AChR抗体阳性MG比较更易发生肌肉束颤、腹泻等胆碱能超敏反应[3,12],本文经案例回顾分析亦发现此类患者对新斯的明的反应欠佳。4例患者肌电图检查未见肢体低频递减,3例表现为面部低频递减,符合MuSK抗体阳性MG的电生理特点。MuSK抗体阳性MG少见肢体远端的重复电刺激异常,肌电图异常多累及面部肌肉,面部肌肉低频递减诊断MuSK-MG的灵敏度达75%~85%,肌电图结果显示的面部肌肉异常和临床表现的面肌无力一致。研究发现,颈部椎旁肌、三角肌、额肌和眼轮匝肌的单纤维肌电图检查对MuSK抗体阳性MG的诊断敏感度高,患者早期即可出现jitter增宽[12-13]。本文案例回顾中2例患者出现jitter异常。针对声带受累的MG患者,可选择完善喉部肌电图检查,用于明确MG的诊断,并排除其他神经肌肉疾病[4]。MuSK抗体检测最常用的方法为放射免疫分析法,放射免疫分析法检测显示MuSK抗体阳性结合典型临床表现即可诊断 MuSK抗体阳性MG[14]。
MuSK抗体阳性MG患者常对乙酰胆碱酯酶抑制剂的反应有限,且常出现肌肉束颤、唾液分泌增加等副作用。静脉用丙种球蛋白治疗MuSK抗体阳性MG患者效果亦不佳,MuSK抗体属于IgG4亚型,由于 IgG4 的构象差异,丙种球蛋白不能与其 Fc 受体区域相结合,无法参与补体激活途径,从而使患者对通过封闭Fc受体机制起治疗作用的丙种球蛋白反应差。本文个案回顾中的4例患者对溴吡斯的明疗效不佳,1例对丙种球蛋白疗效差。临床推荐使用皮质类固醇结合血浆置换治疗MuSK抗体阳性MG[12]。本文结果显示,5例患者采用皮质类固醇治疗,4例均有所改善,另有1例采用血浆置换治疗效果非常显著。近年来,利妥昔单抗在MuSK抗体阳性MG的治疗中显示了良好的效果且安全性高[15],中小剂量的利妥昔单抗也可降低 MuSK 抗体滴度并提供持久的免疫抑制效果[16]。个案回顾中的2例患者采用利妥昔单抗治疗均反应良好。双侧声带麻痹可引起急性喉梗阻并危及生命,因此需要动态观察临床症状,必要时行气管插管或切开挽救患者生命[4-5]。
综上所述,双侧声带麻痹为MuSK抗体阳性MG患者的罕见表现,当患者出现伴疲劳现象的发音困难和喘鸣需要考虑该诊断,应完善喉镜进一步确诊。新斯的明试验阴性不能排除该诊断,需要完善MuSK抗体、重频电刺激和单纤维肌电图检查。治疗上类固醇激素、血浆置换和利妥昔单抗治疗效果好,严重患者需要呼吸机支持治疗。早期诊断和针对性治疗非常重要,以免声带闭合导致呼吸衰竭危及患者生命。
猜你喜欢
中国民间疗法(2021年5期)2021-06-09 09:21:22
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:06:30
国际放射医学核医学杂志(2020年2期)2020-05-30 12:39:56
中国中医急症(2019年10期)2019-05-21 07:20:42
现代检验医学杂志(2016年4期)2016-11-15 02:00:58
现代电生理学杂志(2016年3期)2016-07-10 12:10:32
现代电生理学杂志(2016年4期)2016-07-10 12:02:17
现代电生理学杂志(2015年3期)2015-07-18 11:01:04
现代电生理学杂志(2015年2期)2015-07-18 11:00:59
实用医药杂志(2012年6期)2012-04-21 10:38:42
