
高胰岛素血症/高氨血症综合征一家系的遗传学研究
2023-05-15 13:38321000金华市妇幼保健院儿内科第3病区
浙江医学 2023年7期
321000 金华市妇幼保健院儿内科第3病区
赖盼建 李小兵 王大燕 梅金枝
高胰岛素血症/高氨血症(hyperinsulinism- hyperammonemia,HI/HA)综合征是一种遗传异质性疾病,是引起先天性高胰岛素血症(congenitalhyperinsulinism,CHI)的第2 种常见病因[1-2]。目前已发现14 种相关基因变异会引起CHI,包括ABCC8、KCNJ11、GLUD1、GCK、HADH、SLC16A1、UCP2、HNF4A、HNF1A、HK1、PGM1、PPM2、CACNA1D、FOXA2[3]。不同类型CHI 患儿的临床特征、治疗方案及预后不尽相同[3-6]。HI/HA综合征是GLUD1 基因突变引起的[7]。该基因位于染色体10q23.3 上,编码线粒体谷氨酸脱氢酶(glutamate dehydrogenase,GDH)[7]。GLUD1 基因突变降低了GDH 对三磷酸鸟苷(guanosine triphosphate,GTP)变构抑制的敏感性,从而导致其活性异常增高[7]。HI/HA 综合征的特点是反复发作、症状频繁的低血糖和持续的轻度血氨浓度升高。本研究报道一个HI/HA 综合征家系,通过全外显子组测序(whole-exome sequencing,WES)鉴定出1 个GLUD1[c.1466C>T(p.P489L)]杂合错义突变,并探讨该变异位点所致疾病的临床特点,以期提高对HI/HA 综合征的认识。
1 对象和方法
1.1 对象 先证者,女,11 个月。因“反复低血糖2 个月,抽搐2次”于2020年4月13日至金华市妇幼保健院治疗。测得最低血糖1.89 mmol/L,血胰岛素21.73 mU/L,血C-肽3.75 nmol/L,血氨81 μmol/L,生长激素、胰岛素样生长因子-1、皮质醇、促肾上腺素皮质激素水平均在正常范围内。腹部CT 检查未见胰腺影像学异常。血、尿有机酸代谢检测均正常。脑电图及头颅MRI 检查均未见异常。肝、肾功能检查正常。患儿系足月剖宫产,出生体重3.0 kg,出生身高50 cm,Apgar 评分5、10 min 均为10 分。家族史:先证者父亲喝啤酒后容易诱发低血糖,表现为四肢乏力、头晕,既往认为醉酒现象,未重视;先证者祖母有一次输液时出现低血糖发作,表现为乏力、头晕、面色苍白,当时测血糖2.1 mmol/L,平素偶有低血糖发作表现,补充糖分、进食后能缓解,未治疗。先证者祖母兄长及其女儿也有类似低血糖发作,但家属不愿意提供具体资料,拒绝基因检测。患儿家系图见图1。
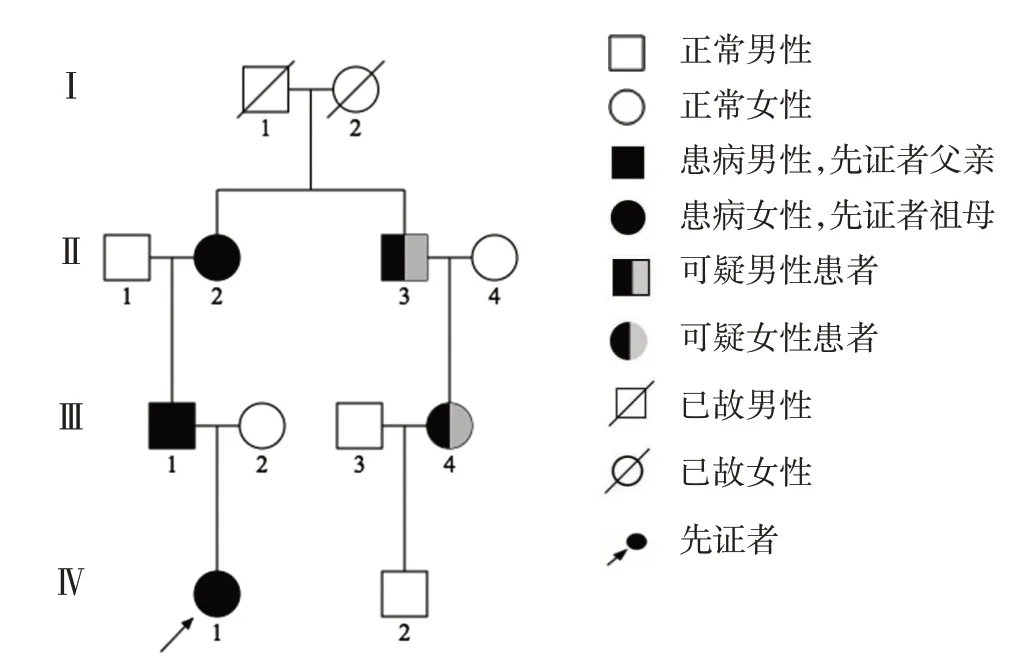
图1 患儿家系图
1.2 DNA 提取 在签署知情同意书后,抽取先证者及其家系成员外周静脉血各2 ml,置入EDTA 抗凝采血管。采用DNA 提取试剂盒[天根生化科技(北京)有限公司]提取血液样本的基因组DNA,并用Qubit 2.0 型荧光计及0.8%琼脂糖凝胶电泳对所提供的DNA 样本进行质检,-20 ℃保存待用。
1.3 人全基因组异常的DNA 拷贝数变异(copy number variation,CNV)检测 利用提取好的DNA 构建DNA 小片段文库,利用Illumina NovaSeq 6000 测序仪(美国Illumina 公司)进行高通量测序,使用Burrows-Wheeler Aligner(BWA)软件将通过质控的DNA 片段与参考序列(参考基因为hg19 基因)进行比对。数据质控通过之后上传智因诊断云平台,对100 kb 及以上区间大小的CNV 进行分析,分析CNV 区间所包括的基因,关联Decipher、ClinVar、OMIM、DGV、ClinGen、ISCA等相关数据库,分析注释已报道的致病片段。
1.4 WES 采用美国IDT 公司的xGen®Exome Research Panel v1.0 捕获探针与gDNA 文库序列进行液体杂交,富集外显子,部分内含子及部分非编码区域,构建整个外显子文库。利用Illumina NovaSeq 6000 测序仪进行高通量测序,目标序列测序覆盖度不低于99%。原始数据经去除接头、低质量测序片段等质控后得到干净数据。使用BWA 软件将测序序列与参考基因hg19 进行比对,使用GATK 软件对单核苷酸多态性和插入缺失突变(indels)(<50 bp)等数据进行分析,并使用SIFT 筛选次要等位基因频率<5%的非同义变异。所有变异和相关疾病均使用dbSNP、1000 基因组项目、ExAC、ESP、OMIM、Swiss var、HGMD、ClinVar SNP等疾病数据库进行注释。此外,Provean、SIFT、Polyphen2-HVAR、Polyphen2-HDIV 和Mutationtster 软件包用于预测所有检测到的变异的功能变化,MaxEntScan软件用于评估剪接位点变异的影响。
1.5 Sanger 测序验证 采用Sanger 测序对先证者及其父母利用全外显子组测序筛选出的候选基因进行验证。引物设计采用Primer 6 软件针对GLUD1 基因c.1466(exon11)C>T(p.P489L)突变设计引物,上游为5'-TCATTGGCTCCTCACTTCCT-3',下游为5'-TTGCCAGTCCAAACCCATG-3',T 值为60 ℃。扩增中所用试剂为Goldstar Taq mix,反应体系为50 μl,使用仪器为ABI PCR扩增仪。PCR反应条件:95 ℃预变性10 min,95 ℃30 s,60 ℃30 s,72 ℃40 s,共30 个循环,最后72 ℃延伸10 min。对所得的PCR 产物进行1%琼脂糖凝胶电泳,进行鉴定,然后对PCR 产物进行一代测序(ABI 3730 一代测序仪),获得数据后采用DNASTAR软件进行序列分析和比对,排除掉二代测序中假阳性的位点。
2 结果
2.1 高通量测序测CNV 的结果 先证者未检测出染色体非整倍体或100 kb以上的已知基因组CNV,见图2。2.2 WES 及Sanger 测序结果 WES 结果显示,先证者存在GLUD1 基因的1 个变异c.1466(exon11)C>T 杂合突变,p.P489L(p.Pro489Leu)(NM_005271)。Sanger 测序结果提示,先证者存在GLUD1 基因c.1466(exon11)C>T杂合突变,其父亲和祖母均存在该突变,而母亲正常,见图3。
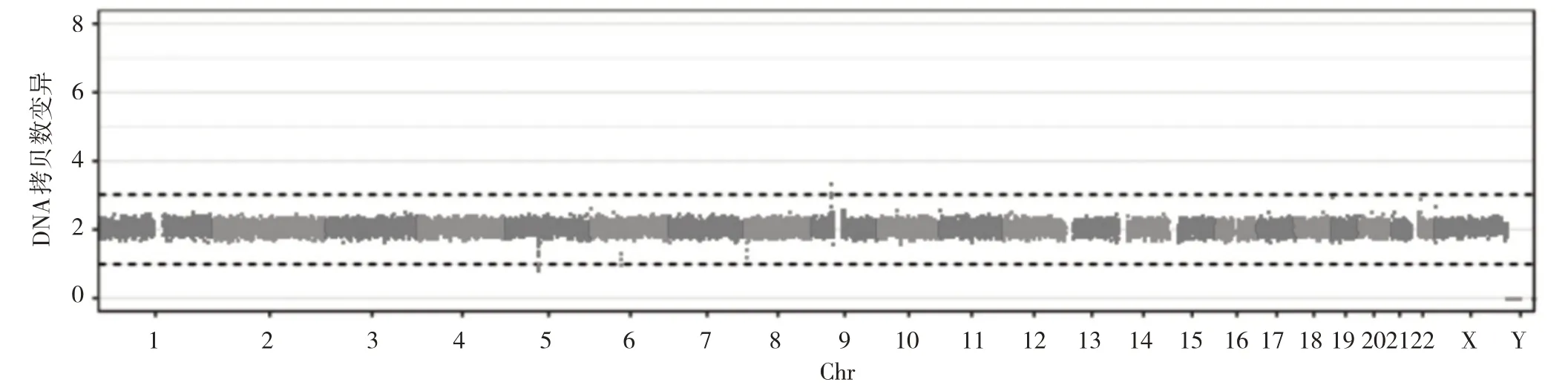
图2 患者DNA 拷贝数变异测序结果
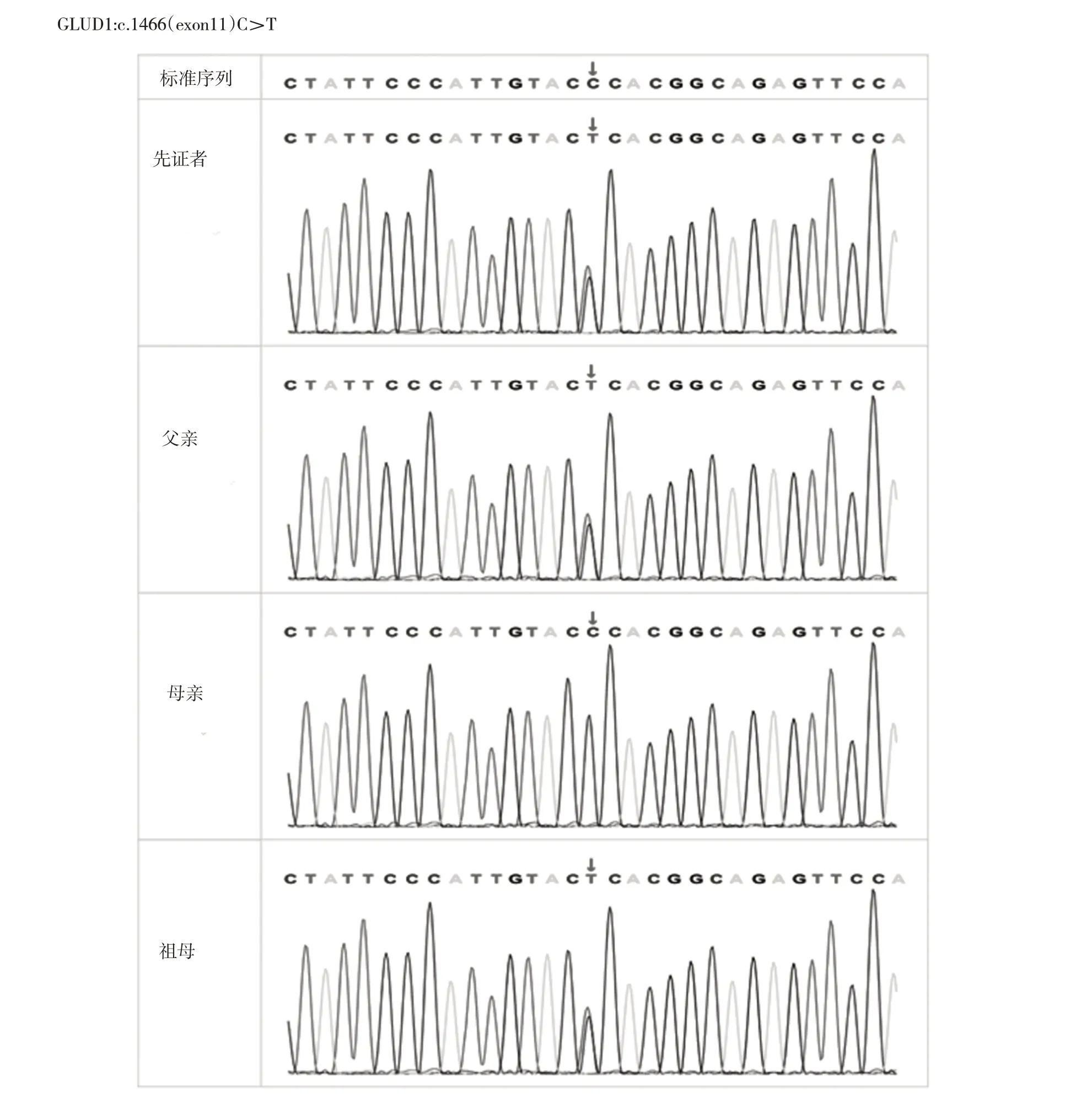
图3 患儿家系GLUD1 基因测序结果
2.3 突变位点致病性分析预测结果 先证者目标区域平均测序深度155.33X,目标区域覆盖度达到99.70%,其中目标区域达到20 以上的区域占全部芯片目标捕获区域的98.67%,芯片对目标区域覆盖良好。经过数据分析发现GLUD1 基因的1 个变异c.1466(exon11)C>T,p.P489L(p.Pro489Leu)(NM_005271)杂合错义突变。生物学致病等级:根据美国医学遗传学与基因组学学会指南(2019 年),变异为可能致病。综合临床分析,最终确定该突变为该家系的可疑致病突变。
2.4 临床治疗及随访 先证者因2 个月内抽搐发作2 次,饮食控制后血糖监测频繁低于3 mmol/L,故加用二氮嗪[5 mg/(kg·d)]口服治疗,空腹血糖维持在3.9 mmol/L 以上,无抽搐发作。父亲和祖母低血糖发作不频繁,予饮食控制后无低血糖发作。祖孙3 人的血生化及血氨基酸检测结果见表1。
3 讨论
GLUD1 基因编码线粒体GDH,它的激活突变引起HI/HA 综合征[7]。GDH 在肝脏、胰腺β 细胞、肾脏和大脑中高度表达[8]。GDH 可以催化谷氨酸的氧化脱氢反应生成a-酮戊二酸和氨。在胰腺β 细胞中,a-酮戊二酸进入三羧酸循环使ATP/二磷酸腺苷(ADP)升高,钾离子通道关闭引起胰岛素释放。GDH 被亮氨酸变构激活,并且被GTP 抑制[9]。GLUD1 基因的火花突变降低了该酶对GTP 变构抑制的敏感性[10]。GTP 抑制作用丧失导致亮氨酸诱导的谷氨酸氧化为a-酮戊二酸,同时肝内GHD 活性增加可引起血氨增加,并破坏尿素循环,使得血氨代谢途径受损,导致血氨升高[11]。因此,高蛋白食物进食后诱发低血糖发作,亮氨酸敏感性是该类疾病的特征。HI/HA 综合征患者的血氨水平常常处于正常水平的3~5 倍,并且很难通过治疗降至正常,持续HA 的机制目前尚未完全了解。
与ABCC8、KCNJ11 基因突变引起的CHI 相比,GLUD1 突变引起的CHI 症状通常较轻,出生时没有巨大儿表现,低血糖发作年龄多在婴儿晚期,大多数患者在2 岁前出现首发症状,平均5~6 个月[12-15]。HI/HA综合征几乎都伴有HA[16],只有少数GLUD1 突变的患者血氨水平没有升高[12,17]。其血氨水平很难通过治疗降至正常,但临床上不会出现HA 的神经系统症状,如嗜睡、呕吐、头痛或急性高氨危象。持续HA 的机制:(1)GDH活性增强,使得谷氨酸产生氨增加;(2)谷氨酸的消耗增多,N-乙酰谷氨酸的产生减少,后者是血氨解毒第一步所必需的变构活化剂[12]。癫痫常被报道与HI/HA综合征有关。文献报道,50%~80%的HI/HA综合征患者会出现其他神经系统表现,如精神发育迟缓和学习障碍[13,18],大多数为癫痫患者。除少数患者外,HI/HA综合征患者的神经影像学表现正常。癫痫的风险与HA 无关,但推测可能是大脑GDH 活性改变导致[19],它导致谷氨酸池的耗竭和谷氨酸与γ-氨基丁酸之间的失衡[13,18]。到目前为止,突变已被描述在GDH的变构区编码氨基酸的外显子6、7、10、11和12中[16]。据报道,癫痫更频繁地与外显子6和7的突变相关[13]。
根据美国内分泌学会2009 年成人低血糖症诊治指南[20],患者血糖<3 mmol/L 时,如同步的胰岛素≥3 mU/L、C-肽≥0.20 nmol/L(0.6 ng/ml)、胰岛素原≥47.17 ng/L(5 pmol/L),考虑为高胰岛素性低血糖症。本组家系3 例患者低血糖发作时,血胰岛素、血C-肽检测均高于正常值,血氨亦超出正常范围,故临床诊断符合HI/HA 综合征。该组家系GLUD1 第11 外显子区域c.1466C>T(p.P489L)杂合错义突变,虽已有相关病例报道[12],但为中国HI/HA 综合征首次报道。Stanley[16]对c.1466(exon11)C>T(p.P489L)位点突变可引起GDH 功能改变进行了验证,结果显示基础GDH 活性正常,但GTP 的半数最大抑制浓度比对照组高470%,提示GDH 活性过高。
综上所述,患儿婴儿期起病,存在低血糖家族史,需警惕CHI,并且对该类患者需常规进行血氨测定,早期基因诊断,早期建立最佳的血糖控制,从而预防以后的神经功能障碍。本组家系同样的基因突变,但是子代低血糖症状比父代重,可能存在家族基因累积效应,有待进一步验证。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
首都食品与医药(2021年5期)2021-03-22
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
转化医学电子杂志(2018年11期)2018-01-16
三门峡职业技术学院学报(2017年1期)2017-06-05
中国洗涤用品工业(2017年2期)2017-04-16
中国比较医学杂志(2017年5期)2017-01-17
中国现代药物应用(2016年10期)2016-03-06
医学研究杂志(2015年12期)2015-06-10
