
基于微卫星的相关性测试对新型冠状病毒的遗传多样性研究
2023-04-12 19:27:07蒋帅游昌乔丁云云张红明秦红郭新红
激光生物学报 2023年3期
蒋帅 游昌乔 丁云云 张红明 秦红 郭新红


摘 要:微衛星(microsatellites)在新型冠状病毒(SARS-CoV-2)的基因组表达调控、种群遗传进化以及宿主免疫互作调节方面发挥重要作用。该研究利用NCBI数据库以及微卫星分析系统筛选并测试SARS-CoV-2原株及变体中不同微卫星数量与遗传特征的关联,探索影响SARS-CoV-2遗传多样性的微卫星特征。通过生物信息学分析,构建SARS-CoV-2的全基因组序列库,并收集关于序列库的遗传特征、微卫星数量以及微卫星相对位置分布特征信息;通过生物统计学分析,对不同微卫星数量进行相关性测试以及单样本Wilcoxon符号秩非参数检验。结果表明,SARS-CoV-2的原株与变体(除Lambda和Omicron)中不同微卫星数量、占比与相对位置分布特征相似。2核苷酸重复(77%~78%)、3次基序重复(22%~23%)以及全长6 bp(73%)的微卫星占比较高,而ORF3a(0.48/100 bp)、E(0.44/100 bp)与N(0.40/100 bp)3个编码区序列的微卫星密度也较高。总平均微卫星数量与碱基含量间存在显著的负相关性(r:–0.799 6;P:0.009 7),但与碱基替换量无显著相关性。本研究丰富了分子生物学领域对SARS-CoV-2的遗传多样性以及进化机制的研究,并为新型冠状病毒感染疫情的防治提供了新思路。
关键词:新型冠状病毒;微卫星;遗传多样性;碱基含量偏向性;种群遗传进化
中图分类号:R373 文献标志码:ADOI:10.3969/j.issn.1007-7146.2023.03.003
Genetic Diversity Analysis of SARS-CoV-2 Based on Correlation Tests of Microsatellites
JIANG Shuai1#, YOU Changqiao1, 2#, DING Yunyun1#, ZHANG Hongming1, 2, QIN Hong2, GUO Xinhong1*
(1. School of Biology, Hunan University, Changsha 410082, China; 2. NanHua Bio-medicine Co., Ltd., Changsha 410006, China)
Abstract: Microsatellites were crucial in the control of SARS-CoV-2 genomic expression, population genetic evolution and host immunity interaction. The NCBI database and microsatellite analysis systems were utilized in this study to screen and test the correlation between the number of different microsatellites and genetic characteristics in the original strain and variants of SARS-CoV-2, as well as to investigate the microsatellite features affecting the genetic diversity of SARS-CoV-2. By bioinformatics analysis, the whole-genome sequence database of SARS-CoV-2 was constructed, and information on the genetic characteristics, microsatellite numbers and relative position distribution characteristics of microsatellites in the sequence database were collected; correlation analysis between the number of various microsatellites and one-sample Wilcoxon signed-rank non-parametric tests were performed by biostatistical analysis. The findings revealed that the quantity, proportion, and relative distributing features of various microsatellites in the original strain and SARS-CoV-2 variants (except Lambda and Omicron) were comparable. The proportion of microsatellites with 2 nucleotide repeats (77%~78%), 3 motif repeats (22%~23%), and 6 bp full-length (73%) were relatively high, and the microsatellite density of ORF3a (0.48/100 bp), E (0.44/100 bp), and N (0.40/100 bp) coding regions were also high. The total average number of microsatellites was negatively correlated with the base content (r: –0.799 6, P: 0.009 7), whereas no significant correlation with the number of base substitutions. This study enriched the research on the genetic diversity as well as the evolutionary mechanism of SARS-CoV-2 in the field of molecular biology and provided new ideas for the prevention and control of the corona virus disease 2019 pandemic.
Key words: SARS-CoV-2; microsatellites; genetic diversity; base content bias; population genetic evolution
(Acta Laser Biology Sinica, 2023, 32(3): 208-216)
作为21世纪以来影响最严重的全球性流行病,新型冠状病毒病(corona virus disease 2019,COVID-19)给世界各国的经济发展和人民的健康生活带来了沉重的影响[1]。新型冠状病毒(severe acute respiratory syndrome coronavirus 2,SARS-CoV-2)是导致此次疫情的致病病原体,属于正义单链RNA病毒(positive-sense single-stranded RNA virus,+ssRNA virus) [2]。自2019年年末,在湖北省武汉市首次检测出SARS-CoV-2后,该毒株及其变体(variants)的全基因组注释[3]、三维蛋白结构[4]以及遗传谱系[5]等信息被逐渐公布。
微卫星(microsatellites)又称简单序列重复(simple sequence repeats),指由1~6 bp长的基序(motifs)串联重复构成的短序列。作为基因组中的重要遗传单元,利用生物信息学手段研究其组成以及分布不仅能够解释生命体的遗传与蛋白表达调控周期特征[6]、重建群落遗传谱系[7],而且有助于促进新疗法的开发[8]。截止到目前,有关对SARS-CoV-2各变体微卫星的分析主要包括2个方面:首先是对微卫星在SARS-CoV-2原株与其他人冠状病毒(human coronaviruses,HCoVs)中的分布信息进行横向种间对比;其次是优化检索和筛选某SARS-CoV-2株系内特定基因中微卫星的算法[9-10]。随着更多SARS-CoV-2变异株的发现与相应的全基因组序列的公开,研究人员往往忽视SARS-CoV-2不同变体之间的微卫星含量与相对位置分布的相关性研究。此外,不同变体的微卫星相关特征与全基因组遗传多样性之间的潜在相关性也存在很大的研究空间。
本文基于SARS-CoV-2原株及其变种全基因组序列内部丰富的遗传多样性特征,利用在线微卫星搜索网站,挖掘、筛选并统计各类微卫星数量及在基因组内部的相对位置分布,通过相关性测试与非参数检验找出与微卫星数量、分布特征相关的遗传学因素并验证结果的准确性,从而对微卫星调控SARS-CoV-2生理活动的潜在新机制进行初步探索与解释。近年来,众多的研究成果已经证明,微卫星的相关研究可以高效准确地揭示病毒与人体之间的互作方式,并针对互作网络中某过程涉及到的部分遗传特征研发特殊抗病性药物[11]。因此,通过分析SARS-CoV-2各变种的遗传多样性与微卫星特征之间的关联,可以进一步挖掘SARS-CoV-2的潜在遗传特征,并为研究人员防治新冠疫情提供新思路。
1 材料与方法
1.1 SARS-CoV-2全基因组序列库构建与比对
出于准确性考虑,从美国国家生物技术信息中心(National Center for Biotechnology Information,NCBI)的Nucleotide数据库[12]中收集SARS-CoV-2原株的全基因組参考序列(complete RefSeq genome,RG)和6种已发表的主要变异株(Alpha B.1.1.7、Belta B.1.351、Delta B.1.617.2、Gamma P.1、Lambda C.37、Omicron B.1.1.529)[2]的全基因组序列,构建fasta格式序列库。简并碱基(degenerate bases,DBs)经常出现在测序质量不佳的基因组中,很容易对开放阅读框处的核酸序列进行错判,且导致部分后续操作软件造成兼容性错误[13]。参考前人对DBs的处理[14],在序列库比对前,利用Python中的re.sub函数批量清除因测序结果不准确造成的基因组序列内部出现的所有DBs(形式包括:RYMKSWHBVDNZ),以避免其影响遗传多样性的分析结果。鉴于序列平均长度较长(约30 000 bp),采用基于MAFFT算法的FFT-NS-1多重序列比对方法对序列库进行在线比对(https://mafft.cbrc.jp/alignment/server/)[15]。
1.2 序列库的生物信息学分析
比对完成后,利用分子进化遗传软件(molecular evolutionary genetics analysis,MEGA11.0)[16]的核苷酸组成查找功能,分析序列库内4种碱基的总平均含量以及3个密码子位点的总平均碱基含量。基于MEGA11.0中的“核苷酸对频率计算”功能,分析序列库所含不同核苷酸对的替换频率分布。过滤后DBs在比对后序列库中形成空位(gaps),将完整的编码序列截断,导致最终搜索到的微卫星数量不准确,各变体相同编码区序列内微卫星的分布情况也难以比较。利用在线微卫星查找工具(simple sequence repeat identification tool,SSRIT,https://archive.gramene.org/db/markers/ssrtool)[17] 在单个序列中查找微卫星存在一定的效率,将选项设置为模糊查找功能,可以降低空位对微卫星的搜索影响。直接采用SSRIT对比对后序列库进行微卫星查找,并设置最大基序长度与最大重复数量参数分别为“trimer”和“3”,以此找出序列库中所有微卫星基序出现重复次数超过3次的2核苷酸重复(dinucleotide repeats,DiRs)、3核苷酸重复(trinucleotide repeats,TriRs)、3次基序重复(triple motif repeats,3MRs)、4次基序重复(quadruple motif repeats,4MRs)以及5次基序重复(quintuple motif repeats,5MRs)的微卫星,同时该平台还标定了所有微卫星的相对起始位置与终止位置区间。
1.3 微卫星的遗传相关性分析
皮尔逊相关系数(Pearson correlation coefficient,r)能够直观、快速反映2个变量的线性相关性的强弱程度。分别统计DiRs、TriRs以及 DiRs + TriRs的微卫星数量,利用r的描述统计方法,将所有碱基组成相同的微卫星总碱基含量(%)作为独立变量与相应的各类微卫星数量(DiRs、TriRs、DiRs + TriRs)分别进行相关性分析(correlation analysis),探索碱基含量对微卫星组成形式的影响。同时,对碱基替换(base substitution)频数[包括碱基转换(base transition)与碱基颠换(base transversion) 2种单核苷酸形式的突变]与全部微卫星以及DiRs之间的数量相关性进行测试。MEGA软件提供的核苷酸对频率查找功能能够获得每种微卫星的碱基替换频数。为避免以上2个相关性分析结果受偶然误差的影响,且不同微卫星数量的分布与SARS-CoV-2遗传特性之间相关性趋势难以估计,因此对上述相关性测试获得的r进行单样本Wilcoxon符号秩非参数检验(P<0.05、零假设中位数=0、备择假设中位数≠0)[18]。通过比较各组非参数检验统计量的精确概率(exact probability of paired samples,EPPS)与渐进概率(asymptotic probability of paired samples,APPS)的大小关系判断相关性结果是否为随机事件[19]。以上相关性测试以及非参数检验通过R语言(psych包与基础函数)完成。
2 结果与分析
2.1 序列库基本信息
通过NCBI数据库的序列筛选与在线多重序列比对,最终得到比对后序列库的基本信息(表1)。比对后序列库内的序列平均长度为29 746 bp,RG和Alpha变体序列相对较长 (>29 900 bp),测序完整度更高;RG、Alpha、Delta、Gamma以及Lambda变体的原序列DBs占比为0,测序结果更稳定,而Belta和Omicron变体序列在比对后序列中平均长度减小,表明原NCBI测序结果中存在少量DBs。鉴于比对后序列库的序列对齐(alignment)特性以及SSRIT网站的自适应算法,由DBs造成的部分序列出现gaps会被清除或用拟合的碱基进行填充。此外,比对前Omicron序列内的DBs经过人工核实多数位于序列末尾,不会影响其余基因编码区内部微卫星的查找与定位。
2.2 序列库内微卫星基本信息
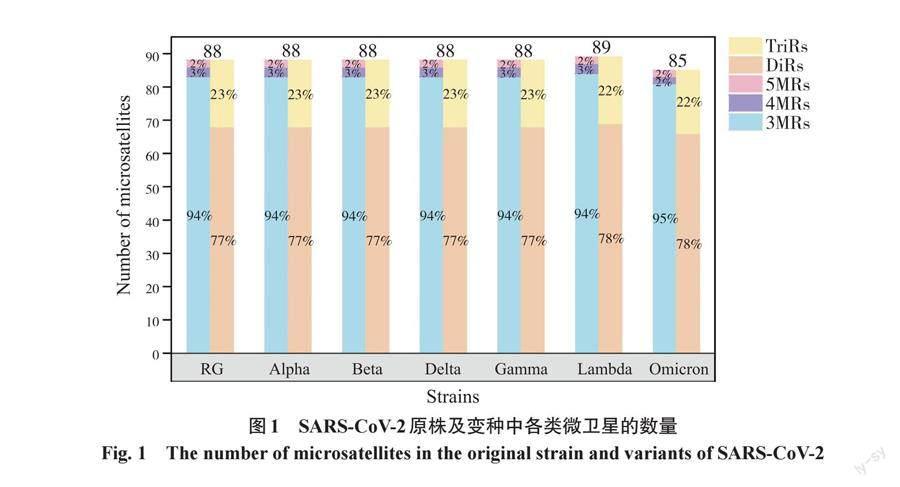
利用SSRIT在线查找比对后序列库内各序列不同种类的微卫星,结果表明,SARS-CoV-2原株及变种中存在相似数量分布特征的各类微卫星(图1)。DiRs和TriRs分别占微卫星总量的77%~78%以及22%~23%;3MRs、4MRs和5MRs则分别占微卫星总量的94%~95%、2%~3%以及2%。基序重复次数越多、基序越复杂的微卫星往往更难在变体中存在。随着更多SARS-CoV-2株系的出现,复杂微卫星数量的改变则暗示了病毒关键位点的突变以及适应环境能力的增强[7]。Lambda和Omicron作为最晚出现的2种变体[20],序列内的微卫星数量与其他变体相比存在一定的差别(89个、85个),且主要体现在DiRs (78%、78%)与TriRs (22%、22%)的微卫星数量所占比例的差异;Omicron变体序列内的3MRs微卫星所占比例较高(95%),说明其内部的特异遗传标记逐渐向生成内部更稳定的结构方向进行变异。此外,DiRs与3MRs在序列内的占比更大且结构更稳定,因此,我们推测以上微卫星可能是促进SARS-CoV-2变种具有丰富遗传多样性的重要因素。
SARS-CoV-2微卫星的长度以6、8、9、10和12 bp为主,6 bp长度的微卫星平均数量超过60个,占总数的73%(图2)。8和10 bp(2个)长度的微卫星数量相似,但数量远小于9 bp长度的微卫星。尽管DiRs的微卫星数量更多,但多以TriRs形式为主,4MRs和5MRs则为罕见形式。10和12 bp长度的微卫星在各变体中的数量相同,6、8和9 bp这类长度较短的微卫星,它们在SARS-CoV-2突变产生不同变体的过程中,已经出现了数量上的变化。Lambda变体和Omicron變体分别在6 bp(65个)以及6、8和9 bp(63个、1个、18个)长度上与其余变体间存在微卫星数量差异。可以假设,随着SARS-CoV-2进一步突变,新的株系中长度较短的微卫星容易先发生变异,而长度较长的微卫星则可以作为病毒内部的保守标记序列检验或预测SARS-CoV-2的突变周期与规模。
以RG序列的基因编码区位置与长度为基准,对齐并录入其余变体的基因编码区相对位置信息,从而整理所有微卫星在各基因编码区的分布密度(图3)。ORF3a(0.48/100 bp),E(0.44/100 bp)与N(0.40/100 bp)3个编码区的微卫星密度最高,其编码区产物分别为ORF3a蛋白[21]、包膜蛋白和核衣壳磷蛋白[22]。不含微卫星的3个编码区分别为ORF6、ORF7b以及ORF8,其编码区产物分别为ORF6蛋白、ORF7b蛋白以及截短ORF8蛋白[21-22]。相较于其他序列的S区内微卫星密度,Omicron变体明显偏小(0.002 4/100 bp),这表明Omicron变体的主要突变定位在S编码区,其产物表面糖蛋白[22]与其他变体间存在一定程度差异。此外,还有3个微卫星未在所有变体的基因编码区出现,它们分别是TG(47~52 bp)、CA(29 507~29 512 bp)以及GT(29 649~29 654 bp)(以RG序列中5'→3'的定位为基准)。
图3 微卫星在SARS-CoV-2原株及变种中各基因编码区的分布
Fig. 3 The distribution of microsatellites in coding regions of the original strain and variants of SARS-CoV-2
A:ORF1ab区;B:S区;C:ORF3a区;D:E区;E:M区;F:ORF6区;G:ORF7a区;H:ORF7b区;I:ORF8区;J:N区;K:ORF10区。
A: ORF1ab region; B: S region; C: ORF3a region; D: E region; E: M region; F: ORF6 region; G: ORF7a region; H: ORF7b region; I: ORF8 region; J: N region; K: ORF10 region.
2.3 碱基含量与微卫星数量的相关性
序列库中的AU碱基对含量(40.0%)明显低于GC碱基对含量(60.0%),相较于一些病毒(如埃博拉病毒[23]),SARS-CoV-2株系存在明显的GC含量偏性,导致该毒株的遗传变异方向存在一定的不稳定性[24](图4)。U-1(12.0%)、G-2(8.1%)、A-2(10.3%)以及C-3(6.9%)分别为4种碱基含量在3个密码子位点的最大占比,表明第1位为U,第2位为G或A,第3位为C的密码子往往在序列中占比更高;A-1(9.5%)、C-1(5.4%)、U-2(8.9%)和G-3(5.1%)则分别为4种碱基含量在3个密码子上的最低占比,表明SARS-CoV-2毒株对第1位为A,第2位为C或U,第3位为G的密码子偏爱程度较低(密码子阅读方向为5'→3')。
依照碱基组成对微卫星归类,共得到9种大类(表2),其中AU/UA/AAU(62.03%)、CGA(67.87%)、CUG/UGC (70.10%)、AUC(80.37%)、AGU/AUG/GAU/UGA(81.67%)的碱基含量均超过60%。相关性测试结果表明,SARS-CoV-2原株及变种内部的碱基含量与微卫星总平均个数之间具有较明显的负相关性(r:– 0.799 6;P:0.009 7),即某类相同碱基组成的微卫星其碱基含量越高,微卫星总平均个数反而越低。同时,非参数检验的配对样本精确概率与渐进概率结果证明,碱基含量与微卫星总平均个数之间的强相关性在SARS-CoV-2中普遍存在,而非偶然出现于某一SARS-CoV-2序列中(0.003 9<0.009 2;r:-0.7996)。对于DiRs或TriRs而言,其总平均个数与碱基含量之间不存在显著相关性(r:0.067 2、P:0.914 5;r:-0.156 3、P:0.688 0)。
圖4 SARS-CoV-2原株及变种中的碱基组成
Fig. 4 The base composition in the original strain and variants of SARS-CoV-2
2.4 碱基替换与微卫星数量的相关性
相同(identical pairs,ii)、转换(transitional pairs,si)以及颠换(transversional pairs,sv)碱基对的总频数分别为29 568、27和16个,而转换与颠换碱基对比值为1.7,表明SARS-CoV-2序列碱基发生替换的主要方式更偏向转换形式(2种嘧啶或2种嘌呤碱基之间的相互突变)。值得注意的是,除所有相同碱基对外,转换(40.8%)和颠换(43.7%)碱基对在密码子第3位(密码子阅读方向为5'→3')的占比最高(图5)。
微卫星总个数(r:-0.540 4、P:0.347 1)和DiRs的总平均个数(r:-0.619 3、P:0.265 3)与SARS-CoV-2原株及变种的碱基转换与颠换数之间均无显著相关性;非参数检验测试也表明,替换的总频数与微卫星的数量间并无显著性差异(表3)。严格来说,碱基替换现象不会直接或间接影响SARS-CoV-2变体内部的微卫星数量发生改变。
3 讨论
微卫星广泛分布于真核生物、原核生物以及病毒的编码区与非编码区[25-26],它们通过调控生物体的基因表达影响物种编码蛋白相关产物的选择以及基因组的进化,目前已成为研究人员分析特定物种遗传多样性的切入点[27]。然而,自2021年11月世界卫生组织正式定义Omicron变体以来,SARS-CoV-2相关的微卫星与遗传多样性的相关性研究一直处于空缺状态[10]。为揭示SARS-CoV-2微卫星对自身遗传多样性的影响,本文首次收集疫情暴发至今所有主要SARS-CoV-2株系的微卫星组成与长度,通过分类学与统计学思路将SARS-CoV-2的微卫星特征与遗传特征的相关性进行多角度剖析,揭示其株系的变异性与多态性规律。文章涉及的物种特异性研究结果有利于为SARS-CoV-2基因组分析、群体研究和物种鉴定提供进一步研究依据;而且类似研究的思路可以改进并推广至其他生物,以揭示各种微卫星在大型基因组序列中的精确分布模式,探索微卫星分布与基因组结构和进化之间的更多联系[28]。
本研究主要采用生物信息学方法,对2019年12月以来的所有SARS-CoV-2重点突变体(重点突变体筛选要求:需考虑社会危害[1]、遗传差异显著性[7]以及数据库注释程度因素[12])的全基因组序列进行分析,获得微卫星数量以及分布信息;并通过生物统计学方法进一步验证了碱基含量与微卫星总量之间具有负相关性而非与序列内存在的碱基替换频数有显著相关性。因此,碱基含量的偏向性往往造成SARS-CoV-2变体内的微卫星数量改变,进而导致更多片段发生突变,造成株系进化的不稳定性与多元性。碱基替换频数与微卫星含量之间总体呈现负相关关系,但单个碱基对的替换变异程度较低且具有偶然性,统计学结果也表明,碱基替换与微卫星含量间无显著差异性(表3)。因此,本文推断,碱基替换量与微卫星含量间仅作为定性关系存在,即替换频数的增长或下降仅导致微卫星含量呈现抑制或上升的趋势。
部分研究者对序列库的构建理念及方法逻辑性一般,泛用性较低[29-31]。本文构建的序列库仅包含NCBI提供的官方参考序列以及已发表的序列,在尽可能确保分析准确性的同时,内部基因注释也为研究微卫星以及遗传多样性在不同基因编码区的位置分布水平提供了参照标准[12]。此外,相比于人体全基因组近1 100个微卫星,病毒基因序列中的微卫星不仅数量少而且长度短[32]。埃博拉病毒(ssRNA virus)基因组序列全长仅为18 900 bp,但序列编码区中共搜索到212个不同种类的微卫星[23],数量近SARS-CoV-2的3倍。因此,相比于其他病毒,SARS-CoV-2自身的遗传信息变化更存在不确定性,免疫逃逸的能力也更强[2]。
周翔等[22]以NCBI的SARS-CoV-2官方参考序列NC_045512为基准,对来自不同国家和地区的36条序列组成的序列库进行了各编码区的单核苷酸的多样性分析。为探索不同编码区功能与微卫星密度的联系,本文参考上述思路设置RG序列为各编码区位置分布基准;并以2019至2022年间出现的重点变体为研究对象,单核苷酸以及微卫星的特征差异为分析手段,从时间角度探索微卫星对SARS-CoV-2的调控潜力以及预测变体可能出现潜在免疫逃逸位点的编码区。ORF3a作为微卫星密度最高的3个编码区之一,其产物ORF3a蛋白属于特异性非结构蛋白,在SARS-CoV-2组装、释放以及炎症应答等方面发挥着关键作用[21]。根据以往SARS-CoV-2全基因组分析,E和S编码区是变异位点出现的集中区域,而N编码区则变异程度相对较低[1, 22]。从功能上看,S蛋白与宿主细胞ACE2受体间表现出较高的亲和力,导致机体更容易出现病症;N和E蛋白均负责病毒的组装,两者的突变均导致SARS-CoV-2致病力的改变[9-10]。ORF6、ORF7b以及ORF8编码区无变异位点且蛋白功能目前尚不清楚[22-23]。因此,ORF3a、E、S和N编码区内的微卫星密度的升降在一定程度上已经开始影响着SARS-CoV-2在致病性和传播能力方面的适应性进化(例如Alpha→Omicron),而这些编码区则往往有高密度的微卫星参与调控。Kirtipal等[2]认为,SARS-CoV-2的进化推动力来自多个编码区的不断变异,针对不同编码区的遗传多样性、系统发育以及编码蛋白结构分析非常有利于研究SARS-CoV-2甚至具有相似遗传特征的HCoVs的遗传进化规律。因此,本文的研究成果还将为其他HCoVs的遗传学研究提供新思路。
碱基含量与变异位点数作为影响微卫星数量的两个潜在因素,部分遗传学研究将它们作为研究物种间遗传变异差异的基础[2, 33-34]。不同物种碱基含量的差异可以直接反映物种间变异能力以及密码子使用偏好的差异,而变异位点数则象征着物种的潜在进化能力[35]。经过改良,我们首次尝试将多种形式的微卫星碱基含量与碱基替换数作为影响微卫星数量的潜在因素,并进行了相关性测试。根据已公布的SARS-CoV-2全基因组的密码子使用模式特征,以U为第1位,A或G为第2位的密码子使用频率较高,但C为第3位的密码子使用频率却很低。同样,以A为第1位,C为第2位或U为第3位的密码子的使用频率较高,但G为第3位的密码子使用频率却很低[33]。对于在密码子第3位GC含量较低的SARS-CoV-2而言,其密码子的使用偏爱程度受突变压力和自然选择影响[33]。此外,SARS-CoV-2和其他HCoVs相似,自身密码子大多数倾向于U结尾,GC含量为30%~40%,且均表现出相似的遗传特性[34]。而高水平的AU碱基对含量(图4中显示为62.0%)能够调节多数影响SARS-CoV-2基因表达的微卫星的稳定性[35],进而导致SARS-CoV-2做出利于自身的突变。转换与颠换为碱基置换(点突变)的主要形式,在具体突变形式未知的前提下[16],将相似碱基组成的微卫星总数与DiRs总平均个数作为变量分别对碱基置换频数进行了相关性测试,最后发现两者间存在不顯著的负相关趋势[35]。
本文部分研究方法的准确性与结论完整性仍需改进。首先,NCBI数据库内上传的全基因组序列中存在部分DBs(表1),导致部分序列的碱基含量和替换数相比于真实情况存在较小误差。其次,本文尚未确定各种微卫星对于SARS-CoV-2的突变是否起到促进或抑制作用,仅能证明其参与基因调控与表达,并可以用作遗传标记物。尽管 DBs对研究结果可能带来一定的影响,但正如“2.1 序列库基本信息”部分所述,本文尽可能采用合适的算法弥补该缺陷,人工查找DBs所在编码区域以确保其不会对整体数据造成严重影响,并列出了微卫星在各基因编码区的分布情况(图3)以证明部分DBs不会对微卫星的数量统计造成较大影响;而比对后序列库的平均碱基含量、微卫星总平均个数以及平均替换数作为研究相关性的因素也不会因为少量DBs产生严重误差。未来,我们会深入研究微卫星相关特征与其他可能影响SARS-CoV-2的遗传特性因素间的联系,进一步完善技术流程,揭示微卫星调控SARS-CoV-2遗传变异与结构功能等的生物学意义,深化研究人员对SARS-CoV-2的认识,获得更有学术意义或应用价值的结果。
参考文献(References):
[1] MORSE S S, MAZET J A, WOOLHOUSE M, et al. Prediction and prevention of the next pandemic zoonosis[J]. Lancet, 2012, 380(9857): 1956-1965.
[2] KIRTIPAL N, BHARADWAJ S, KANG S G. From SARS to SARS-CoV-2, insights on structure, pathogenicity and immunity aspects of pandemic human coronaviruses[J]. Infection Genetics and Evolution, 2020, 85: 104502.
[3] NIMAVAT N, SINGH S, FICHADIYA N, et al. Online medical education in India-different challenges and probable solutions in the age of COVID-19[J]. Advances in Medical Education and Practice, 2021, 12: 237-243.
[4] SWANSON S J, CONANT L L, HUMPHRIES C J, et al. Changes in description naming for common and proper nouns after left anterior temporal lobectomy[J]. Epilepsy & Behavior, 2020, 106: 106912.
[5] PENG C, HE M, CUTRONA S L, et al. Theme trends and knowledge structure on mobile health APPS: bibliometric analysis[J]. JMIR mHealth and uHealth, 2020, 8(7): e18212.
[6] KIM T S, BOOTH J G, GAUCH H G, et al. Simple sequence repeats in Neurospora crassa: distribution, polymorphism and evolutionary inference[J]. BMC Genomics, 2008, 9: 31-50.
[7] FLANAGAN S P, JONES A G. The future of parentage analysis: from microsatellites to SNPs and beyond[J]. Molecular Ecology, 2019, 28(3): 544-567.
[8] BOUDREAU A, RICHARD A J, HARVEY I, et al. Artemisia scoparia and metabolic health: untapped potential of an ancient remedy for modern use[J]. Frontiers in Endocrinology, 2022, 12: 727061.
[9] SAVARI H, SHAFIEY H, SAVADI A, et al. Statistics and patterns of occurrence of simple tandem repeats in SARS-CoV-1 and SARS-CoV-2 genomic data[J]. Data in Brief, 2021, 36: 107057.
[10] REHMAN H A, RAMZAN F, BASHARAT Z, et al. Comprehensive comparative genomic and microsatellite analysis of SARS, MERS, BAT-SARS, and COVID-19 coronaviruses[J]. Journal of Medical Virology, 2021, 93(7): 4382-4391.
[11] LIN C Y, MEHTA P, WATERS K M, et al. Complete response to neoadjuvant pembrolizumab and capecitabine in microsatellite stable, Epstein-Barr virus-positive, locally advanced gastric adenocarcinoma: case report[J]. American Journal of Case Reports, 2021, 5: 30.
[12] SCHOCH C L, CIUFO S, DOMRACHEV M, et al. NCBI taxonomy: a comprehensive update on curation, resources and tools[J]. Database (Oxford), 2020, 2020: baaa062.
[13] 鐘东. DNA序列的对称性与真核基因调控元件模块的分析[D]. 广州: 南方医科大学, 2003.
ZHONG Dong. Symmetry in DNA and analysis of eukaryotic cis-regulatory module[D]. Guangzhou: Southern Medical University, 2003.
[14] LINHART C, SHAMIR R. The degenerate primer design problem: theory and applications[J]. Journal of Computational Biology, 2005, 12(4): 431-456.
[15] ROZEWICKI J, LI S, AMADA K M, et al. MAFFT-DASH: integrated protein sequence and structural alignment[J]. Nucleic Acids Research, 2019, 47(W1): W5-W10.
[16] TAMURA K, STECHER G, KUMAR S. MEGA11: molecular evolutionary genetics analysis version 11[J]. Molecular Biology and Evolution, 2021, 38(7): 3022-3027.
[17] YOUENS-CLARK K, BUCKLER E, CASSTEVENS T, et al. Gramene database in 2010: updates and extensions[J]. Nucleic Acids Research, 2011, 39 (Database issue): D1085-D1094.
[18] BORGIA E, BARON R, BORGIA J L. Quality and survival of direct light-activated composite resin restorations in posterior teeth: a 5- to 20-year retrospective longitudinal study[J]. Journal of Prosthodontics-Implant Esthetic and Reconstructive Dentistry, 2019, 28(1): e195-e203.
[19] DIVINE G, NORTON H J, HUNT R, et al. Statistical grand rounds: a review of analysis and sample size calculation considerations for Wilcoxon tests[J]. Anesthesia and Analgesia, 2013, 117(3): 699-710.
[20] WOLFE M, HUGHES B, DUONG D, et al. Detection of SARS-CoV-2 variants Mu, Beta, Gamma, Lambda, Delta, Alpha, and Omicron in wastewater settled solids using mutation-specific assays is associated with regional detection of variants in clinical samples[J]. Applied and Environmental Microbiology, 2022, 88(8): e0004522.
[21] 高文欣, 李希琳, 傅煜軒. 新型冠状病毒辅助蛋白ORF3a、ORF3b的致病机制研究[J]. 实用临床医药杂志, 2022, 26(11): 1-5.
GAO Wenxin, LI Xilin, FU Yuxuan. Pathogenic mechanism of severe acute respiratory syndrome coronavirus 2 protein ORF3a and ORF3b[J]. Journal of Clinical Medicine in Practice, 2022, 26(11): 1-5.
[22] 周翔, 赵仁生, 崔艺璇, 等. SARS-CoV-2病毒全基因组序列比对及进化分析[J]. 云南民族大学学报 (自然科学版), 2022, 31(2): 176-185.
ZHOU Xiang, ZHAO Rensheng, CUI Yixuan, et al. Sequence alignment and evolutionary analysis of SARS-CoV-2 virus[J]. Journal of Yunnan Minzu University (Natural Sciences Edition), 2022, 31(2): 176-185.
[23] LI D, SHI R, ZHANG H, et al. The only conserved microsatellite in coding regions of ebolavirus is the editing site[J]. Biochemical and Biophysical Research Communications, 2021, 565: 79-84.
[24] KOGAY R, WOLF Y I, KOONIN E V, et al. Selection for reducing energy cost of protein production drives the GC content and amino acid composition bias in gene transfer agents [J]. mBio, 2020, 11(4): e01206- e01220.
[25] BAGSHAW A T, PITT J P, GEMMELL N J. High frequency of microsatellites in S. cerevisiae meiotic recombination hotspots[J]. BMC Genomics, 2008, 9: 49.
[26] LOIRE E, HIGUET D, NETTER P, et al. Evolution of coding microsatellites in primate genomes[J]. Genome Biology and Evolution, 2013, 5(2): 283-295.
[27] ZENG L, CHEN N, YAO Y, et al. Analysis of genetic diversity and structure of Guanzhong horse using microsatellite markers[J]. Animal Biotechnology, 2019, 30(1): 95-98.
[28] MUTI H S, HEIJ L R, KELLER G, et al. Development and validation of deep learning classifiers to detect Epstein-Barr virus and microsatellite instability status in gastric cancer: a retrospective multicentre cohort study[J]. The Lancet Digital Health, 2021, 3(10): e654-e664.
[29] LI H, XIAO W, TONG T, et al. The specific DNA barcodes based on chloroplast genes for species identification of Orchidaceae plants[J]. Scientific Reports, 2021, 11(1): 1424.
[30] MAHIMA K, SUNIL KUMAR K N, RAKHESH K V, et al. Advancements and future prospective of DNA barcodes in the herbal drug industry[J]. Frontiers in Pharmacology, 2022, 13: 947512.
[31] GUO Y Y, HUANG L Q, LIU Z J, et al. Promise and challenge of DNA barcoding in Venus slipper (Paphiopedilum)[J]. PLoS One, 2016, 11(1): e0146880.
[32] WEBSTER M T, SMITH N G, ELLEGREN H. Microsatellite evolution inferred from human-chimpanzee genomic sequence alignments[J]. Proceedings of the National Academy of Sciences of the United States of America, 2002, 99(13): 8748-8753.
[33] HOU W. Characterization of codon usage pattern in SARS-CoV-2[J]. Virology Journal, 2020, 17(1): 138-148.
[34] DILUCCA M, FORCELLONI S, GEORGAKILAS A G, et al. Codon usage and phenotypic divergences of SARS-CoV-2 genes[J]. Viruses, 2020, 12(5): 498-519.
[35] JIANG S, CHEN F, QIN P, et al. The specific DNA barcodes based on chloroplast genes for species identification of Theaceae plants[J]. Physiology and Molecular Biology of Plants, 2022, 28(4): 837-848.
猜你喜欢
特产研究(2022年6期)2023-01-17 05:05:06
上海医药(2020年3期)2020-03-23 09:35:30
会计之友(2020年6期)2020-03-11 13:18:22
环球时报(2020-01-10)2020-01-10 04:16:02
大经贸(2019年12期)2019-03-09 11:29:39
四川动物(2017年4期)2017-07-31 23:54:19
长江蔬菜·学术版(2016年12期)2017-01-12 20:57:59
中国科技博览(2016年25期)2016-12-20 20:04:30
农家科技下旬刊(2016年9期)2016-12-15 10:42:25
中国科技博览(2016年11期)2016-05-06 02:35:39
