
反相高效液相色谱法测定抗体药物中组氨酸的含量
2023-03-22 01:01杜加亮梅玉婷于传飞中国食品药品检定研究院单克隆抗体产品室卫生部生物技术产品检定及标准化重点实验室国家药品监督管理局生物制品质量研究与评价重点实验室北京102629共同第一作者通讯作者mailwanglannifdcorgcn
山西医科大学学报 2023年2期
杜加亮,武 刚,梅玉婷,于传飞,王 兰(中国食品药品检定研究院单克隆抗体产品室,卫生部生物技术产品检定及标准化重点实验室,国家药品监督管理局生物制品质量研究与评价重点实验室,北京 102629;共同第一作者;通讯作者,E-mail:wanglan@nifdc.org.cn)
采用高蛋白浓度(≥100 mg/ml)配制的治疗性抗体药物约占美国食品药品管理局(FDA)批准的全部治疗性抗体药物的三分之一(34/103),而且2015年以后批准的数量占76%(26/34),最高浓度为200 mg/ml,主要为皮下给药途径[1]。抗体药物的高浓度制剂将是治疗性抗体制剂发展的未来趋势[2]。但是,高浓度抗体药物的开发也面临着许多挑战,其中辅料配方可以影响药物的稳定性(聚体、氧化等)[3]。研究表明,使用包含聚山梨醇酯、组氨酸和蔗糖的平台制剂以加速抗体药物的高浓度制剂开发可能是合理的[1]。在FDA批准的34种治疗性抗体高浓度制剂的辅料中,聚山梨酯(94%)、组氨酸(82%)、蔗糖(48%)是使用频率排名前三的辅料[1,4]。因此,在高浓度抗体药物放行时,要对辅料进行相应的质量控制。
聚山梨酯是最常用的表面活性剂,而组氨酸有助于聚山梨酯颗粒的稳定性以及维持治疗性蛋白制品的pH值[5-7]。组氨酸因不含生色基团,无法采用常规的紫外检测器直接检测,所以需要通过衍生剂将氨基酸转化为具有较强紫外或荧光吸收的衍生物[8]才能被检测到。本研究拟采用柱前衍生反相高效液相色谱法(reverse-phase high-performance liquid chromatography,RP-HPLC)建立组氨酸含量测定方法并进行全面的方法学验证。
1 材料与方法
1.1 药品及试剂
单抗药物样品mAb-1(蛋白浓度为4.5 mg/ml)为含有已知浓度L-组氨酸(理论浓度为3.10 mg/ml)、蔗糖和吐温-20为辅料的单抗样品,由本实验室保存。L-组氨酸购自美国Sigma公司,符合英国药典、欧洲药典、美国药典/国家处方集和日本药典标准,批号为H6034。蔗糖购自德国Millipore公司(货号:100892),吐温-20购自德国Fluka公司(货号:44112)。色谱级乙腈购自美国Sigma公司(货号:34851)。AccQTag洗脱剂A(货号:WAT052890)和AccQ Fluor试剂盒(货号:WAT052880)购自美国waters公司。
1.2 高效液相色谱系统及参数
使用配有紫外检测器和数据采集系统的Waters HPLC系统Alliance e2695和氨基酸色谱柱(AccQ Tag,粒径4 μm,3.9 mm×150 mm)。色谱条件如下:流动相A为用水1 ∶10(V/V)稀释的AccQTag洗脱剂A,流动相B为含60%乙腈的水溶液。紫外检测器检测波长为254 nm,进样体积为5 μl,柱温为25 ℃,样品室温度5 ℃,流速为1.0 ml/min,洗脱时间为35 min,洗脱方式为梯度洗脱(0~26 min,4%→7%流动相B;26~26.01 min,7%→100%流动相B;26.01~30 min,100%→4%流动相B;30~35 min,4%流动相B)。
1.3 样品制备
用超纯水配制两份单独的浓度为0.03 mg/ml的组氨酸溶液,作为组氨酸标准溶液1和2(His_STD-1和His_STD-2),其中His_STD_1用于样品中组氨酸浓度计算和系统适应性计算,His_STD_2用做质控样品。空白样品为超纯水。专属性样品为配方缓冲液1(含5%蔗糖、0.01%吐温20、20 mmol/L组氨酸,pH 6.0)和不含组氨酸的配方缓冲液(含5%蔗糖、0.01%吐温20,pH 6.0)。使用不含组氨酸的配方缓冲液(也用作加标缓冲液1)和含组氨酸的配方缓冲液2(含15.5 mg/ml组氨酸、5%蔗糖、0.01%吐温20,pH 6.0)分别按照9 ∶1,8 ∶2,7 ∶3和6 ∶4的体积比例配制组氨酸理论浓度分别为1.55,3.10,4.65,6.21 mg/ml的加标缓冲液2~5,然后分别混合1 400 μl mAb-1抗体溶液和600 μl加标缓冲液1~5,配制成组氨酸理论浓度分别为2.170,2.635,3.100,3.565,4.033 mg/ml的70%,85%,100%,115%和130%的加标溶液1~5(见表1),用于准确性、线性和检测范围的验证。

表1 加标溶液的制备Table 1 Preparation of the spiking solutions
1.4 样品进样与数据分析
首先用超纯水平衡系统,然后再依次按以下顺序进样:组氨酸标准品1(6次)、组氨酸标准品2(2次)、样品(2次)、组氨酸标准品1(1次)。组氨酸标准品的括号进样(序列前6次和序列后1次)用于评价系统适应性,且进样次数足够用于统计学分析。样品重复进样2次取平均值用于计算样品中组氨酸浓度。选取色谱图中的组氨酸做积分运算,使用组氨酸标准品1的浓度与峰面积绘制经过原点的直线方程:组氨酸标准品1平均峰面积=b(斜率)×组氨酸标准品1浓度,则样品中组氨酸的浓度(mg/ml)=样品峰面积×100÷b=样品峰面积×组氨酸标准品1浓度×100÷组氨酸标准品1平均峰面积(其中100为样品的稀释倍数)。
1.5 验证方案
1.5.1 专属性 使用mAb-1和不含组氨酸的配方缓冲液进行专属性验证,分别进样3次,可接受标准为在组氨酸的保留时间范围内必须无配方缓冲液组分和分析溶剂的干扰,且mAb-1和组氨酸标准品溶液中组氨酸保留时间的偏差必须≤5%。
1.5.2 准确度 使用加标溶液70%,100%和130%进行准确度验证,可接受标准为各水平的平均回收率为90%~110%,且各水平下3次独立测定的浓度变异系数必须≤5%。
1.5.3 重复性 使用mAb-1进行重复性验证,可接受标准为6次独立测定的浓度变异系数必须≤5%。
1.5.4 中间精密度 由两位实验人员分别在3个工作日对mAb-1进行3次独立的组氨酸含量测定用于评估中间精密度,可接受标准为18次独立测定的浓度变异系数必须≤10%。
1.5.5 线性关系 使用加标溶液1~5进行线性验证。以组氨酸含量为横坐标,峰面积为纵坐标绘制回归方程。可接受标准为决定系数R2≥0.98,y轴截距的置信区间必须包含原点,各浓度水平的回收率应在90%~110%范围内,变异系数应≤5%。
1.5.6 检测范围 用检测范围下限(70%加标)和上限(130%加标)的6次测定验证检测范围,可接受标准为各水平的平均回收率必须在90%~110%范围内,各水平的变异系数必须≤5%。
1.5.7 耐用性 样品和对照品于自动进样器中贮存0 h和24 h后,在同一个系统上分析样品,评价各自的稳定性,可接受标准为样品溶液、标准溶液和对照品溶液的各时间点的平均回收率与0 h相比必须在90%~110%范围内。
2 结果
2.1 RP-HPLC法测定单抗药物中组氨酸含量的专属性验证
结果表明所有配方缓冲液组分均不干扰组氨酸峰(见图1),mAb-1样品和组氨酸标准品1中组氨酸的保留时间(分别为17.789,17.870 min)偏差为0.45%(见图2),符合验收标准,表明该方法具有良好的专属性。
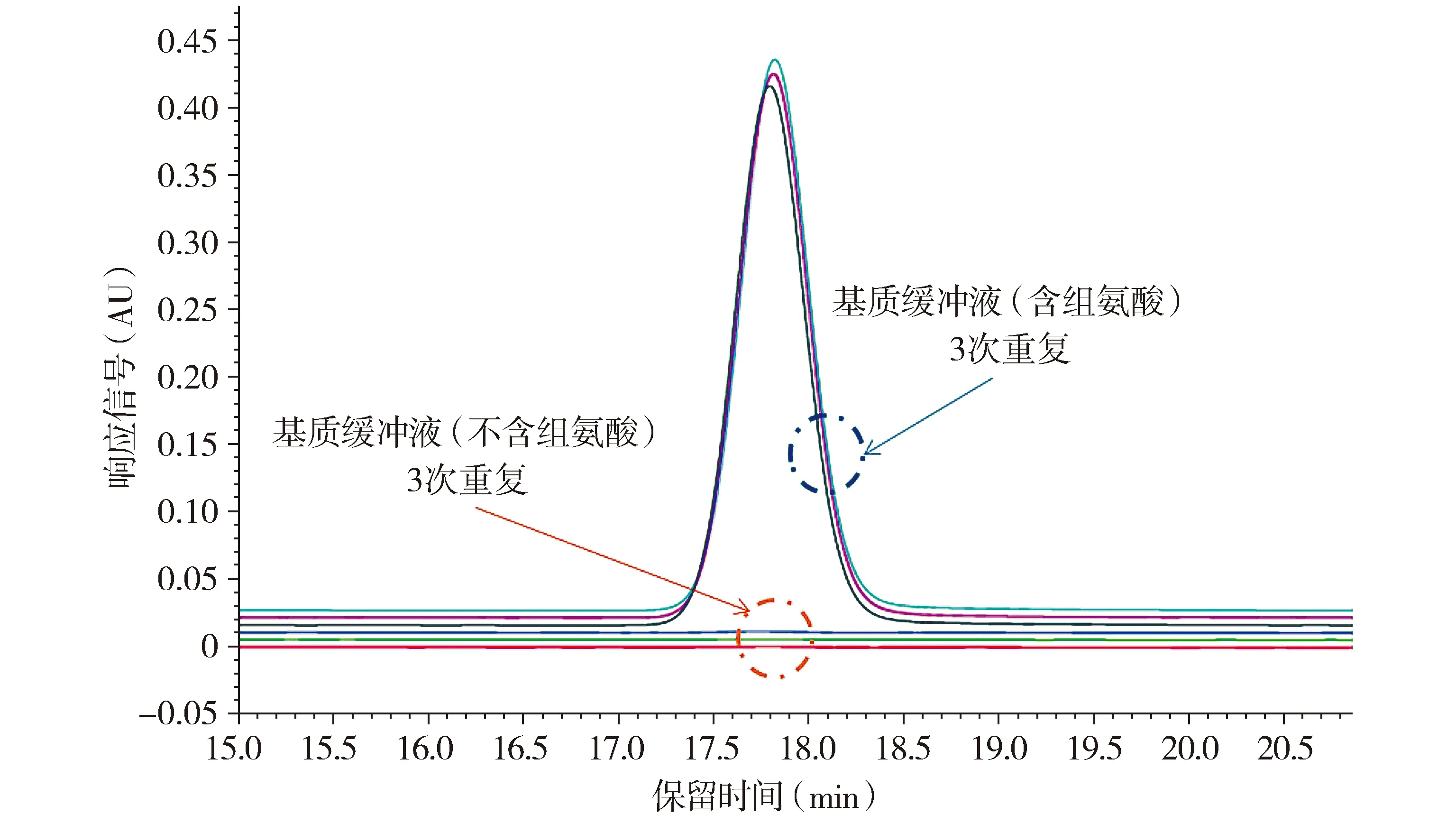
图1 基质缓冲液和不含组氨酸的基质缓冲液的重叠色谱图Figure 1 Overlaid chromatograms of matrix buffer and matrix buffer without histidine

图2 组氨酸标准品、mAb-1和对照品的重叠色谱图Figure 2 Overlaid chromatograms of histidine standard, mAb-1 and blank samples
2.2 RP-HPLC法测定单抗药物中组氨酸含量的准确度验证
结果表明3个加标溶液70%,100%,130%的平均回收率在96.1%~101.9%范围内,变异系数在0.5%~1.1%范围内(见表2),符合验收标准,表明该方法的准确度符合要求。

表2 RP-HPLC法测定单抗药物中组氨酸含量的准确度实验结果Table 2 Results on the accuracy of RP-HPLC method for determining histidine content in monoclonal antibody drugs
2.3 RP-HPLC法测定单抗药物中组氨酸含量的重复性(精密度)验证
重复性验证使用的mAb-1(蛋白浓度为4.5 mg/ml)为含有已知浓度L-组氨酸(理论浓度为3.100 mg/ml)的单抗溶液。6次独立测定结果显示,组氨酸的实测浓度均值为(3.151±0.038)mg/ml,变异系数为1.2%,95%置信区间为3.110~3.191 mg/ml(见表3),符合验收标准,表明该方法具有很好的重复性。

表3 RP-HPLC法测定单抗药物中组氨酸含量的重复性实验结果Table 3 Results on the repeatability of RP-HPLC method for determining histidine content in monoclonal antibody drugs
2.4 RP-HPLC法测定单抗药物中组氨酸含量的中间精密度验证
中间精密度验证使用的样品同重复性验证。两位实验人员分别在3个工作日进行的18次独立测定结果显示,组氨酸平均浓度为(3.273±0.101)mg/ml,变异系数为3.1%,95%置信区间为3.167~3.379 mg/ml(见表4),符合验收标准,表明该方法通过了中间精密度验证的要求。

表4 RP-HPLC法测定单抗药物中组氨酸含量的中间精密度实验结果Table 4 Results on the intermediate precision of RP-HPLC method for determining histidine content in monoclonal antibody drugs
2.5 RP-HPLC法测定单抗药物中组氨酸含量的线性验证
线性验证使用的是5个不同水平的加标溶液,每个浓度点进行3次独立测试,以组氨酸计算浓度为横坐标,以峰面积为纵坐标绘制散点图和趋势线。结果显示,组氨酸浓度和峰面积形成y=109 274x-6 925.8的线性关系,其决定系数R2=0.995 0(见图3)。y轴截距的95%置信区间(-3 328.6~9 262.3)包含原点。各水平下的回收率和变异系数均在验收标准范围内(见表5),表明在分析范围内(2.170~4.033 mg/ml,即10.85~20.15 μg)组氨酸浓度与检测响应信号呈线性关系。

表5 RP-HPLC法测定单抗药物中组氨酸含量的线性实验结果Table 5 Results on the linearity of RP-HPLC method for determining histidine content in monoclonal antibody drugs

图3 组氨酸浓度与检测信号的线性关系Figure 3 Linear correlation of histidine concentration and detection signals
2.6 RP-HPLC法测定单抗药物中组氨酸含量的检测范围验证
本部分再次对组成线性的上限和下限2个浓度点分别进行了6次独立的测试。结果证明,检测范围的下限(平均回收率:94.2%/CV=1.1%)和上限(平均回收率:95.1%/CV=1.1%)均具有良好的精密性和准确度(见表6),符合验收标准。

表6 RP-HPLC法测定单抗药物中组氨酸含量的检测范围实验结果Table 6 Results on the detection range of RP-HPLC method for determining histidine content in monoclonal antibody drugs
2.7 RP-HPLC法测定单抗药物中组氨酸含量的耐用性验证
该方法中,对结果影响较大的因素为衍生后样品的稳定性,因此耐用性主要考察该影响因素。结果表明,相对于0 h,衍生后mAb-1样品在(5±3)℃密封瓶条件下在自动进样器中暂存24 h后的回收率为100.3%,而衍生对照品在(5±3)℃密封瓶条件下在自动进样器中暂存26 h后的回收率为100.2%(见表7),均符合验收标准。

表7 RP-HPLC法测定单抗药物中组氨酸含量的耐用性实验结果Table 7 Results on the robustness of RP-HPLC method for determining histidine content in monoclonal antibody drugs
3 讨论
由于多数氨基酸没有显色基团,无法用简单的方法检测,而且所有的氨基酸极性都很大,无法反相分离。因此需要进行衍生处理,增加显色基团(紫外或荧光),提高检测灵敏度,同时改变样品的极性,变离子型化合物为非离子型,改变分离模式,用反相方法分离[9]。本研究利用6-氨基喹啉-N-羟基琥珀酰亚胺基氨基甲酸酯(6-aminoquinolyl-N-hydroxysuccinimidyl-carbamate,AQC)衍生试剂建立的柱前衍生RP-HPLC紫外检测单抗药物中组氨酸含量的方法在2.17~4.03 mg/ml(即10.85~20.15 μg)范围内,进样量与峰面积呈良好的线性关系。同时,本研究严格按照《中国药典》2020年版三部中分析方法验证指导原则[10]和ICH协调三方指导原则(ICH Harmonised Tripartite Guideline)Q2[11]进行方法学验证。
理论上,方法验证中应使用除了不含待测物质外,其他成分与待测样本完全一致的溶液来评价其专属性[7,12,13]。但是,在单抗的生产工艺中,为了保证原液的稳定性,在原液制备时就加入了配方缓冲液。在此之前的中间产物虽然不含配方缓冲液,但是不易获取和保存。首先通过使用不含组氨酸的配方缓冲液和完整配方缓冲液的比较,排除其他辅料对组氨酸检测结果的影响。在此基础上,通过使用含完整配方的抗体溶液和组氨酸标准品保留时间的比较,检查抗体蛋白对组氨酸的保留时间是否有影响。
准确度验证使用了高(4.033 mg/ml)、中(3.100 mg/ml)、低(2.170 mg/ml)3个浓度的组氨酸加标溶液。如上所述,由于无法获得不含组氨酸的抗体溶液,本研究中使用的加标溶液(也包括文中线性和工作范围验证中使用到的加标溶液)是使用含完整配方的抗体溶液和含不同浓度组氨酸的配方缓冲液按一定比例配制而成,以其理论浓度作为真实值,比较检测结果与真实值的偏差。
在线性验证中,组氨酸浓度和峰面积可以形成一条经过原点的直线方程。因此,在方法的实际应用时,为了简化试验操作和计算过程,使用单个点的组氨酸标准品浓度与峰面积绘制经过原点的直线方程,得到峰面积相对于浓度的斜率,再通过样品的峰面积和斜率计算样品中组氨酸的浓度。
综上所述,该方法具有良好的专属性、准确度、重复性、中间精密度、线性和耐用性,可作为单克隆抗体制品的组氨酸质量控制方法。
猜你喜欢
河北北方学院学报(自然科学版)(2022年11期)2022-02-03
食品与发酵工业(2021年12期)2021-07-05
中国科技纵横(2021年24期)2021-03-02
昆钢科技(2020年6期)2020-03-29
中国蔬菜(2019年4期)2019-06-06
中成药(2018年6期)2018-07-11
中国饲料(2018年7期)2018-01-24
中成药(2017年9期)2017-12-19
中成药(2017年5期)2017-06-13
中成药(2016年8期)2016-05-17
