
THR β基因c.728G>A突变致甲状腺激素抵抗综合征家系6年随访
2023-03-06 10:09郭曼丽仇亚丽李京马绍刚
温州医科大学学报 2023年2期
郭曼丽,仇亚丽,李京,马绍刚
1.徐州医科大学附属宿迁医院 南京鼓楼医院集团宿迁医院 内分泌科,江苏 宿迁 223800;2.宿迁市妇幼保健院 新生儿疾病筛查中心,江苏 宿迁 223800;3.宿迁市第一人民医院 内分泌科,江苏 宿迁 223800;4.蚌埠市第三人民医院 内分泌科,安徽 蚌埠 233000
甲状腺激素抵抗综合征(resistance to thyroid hormone syndrome,RTH)是一种以机体组织器官对甲状腺激素敏感性降低为特点的罕见遗传病,发病率为1∶(40 000~50 000),其血清学特点为升高的甲状腺激素水平和不被抑制的促甲状腺激素(thyroid stimulating hormone,TSH)。研究显示,RTH与甲状腺激素受体(thyroid hormone receptor,THR)、单羧酸转运体8、硒代半胱氨酸插入序列结合蛋白2突变有关,其中以THR突变最为常见[1]。THR包括THR α和THR β两个亚型。THR β亚基由位于3号染色体上的THRβ基因编码。THRβ基因突变是RTH最常见的病因,称为RTH β。目前报道的突变大部分为错义突变,多位于3个热点区:310-353(cluster 1),429-461(cluster 2),234-282(cluster 3)[2-3]。THRβ基因突变大部分为常染色体显性遗传,仅见1例报道为常染色体隐性遗传[4-5]。由于靶组织THR对甲状腺激素抵抗的严重程度不同,RTH β患者临床表型异质性大,可以为甲亢、甲减表现或无任何症状,大部分为无症状者,临床中容易造成漏诊或者误诊而给予不适当治疗。因此,分子遗传学在RTH诊断和准确分类中具有重要价值。
迄今为止,国内外已报道了3 000多例RTH患者,100多个THRβ基因突变位点[6]。国内对于RTH遗传学研究相对较少,报道病例不足150例,以散发病例为主,突变位点多位于第9-10外显子[7-8]。笔者报告1个罕见突变位点所致的RTH家系并进行6年随访研究,以提高临床医师对RTH的认识。
1 对象和方法
1.1 对象 先证者出生于2015年12月,足月产,出生时体质量3.0 kg,身长50.0 cm,Apgar评分9分。新生儿筛查测定干血滤纸片TSH值为14.7 μIU/mL(参考值范围:<9 μIU/mL),2 周后复筛TSH 22.77 μIU/mL。出生1个月后患儿被召回至当地医院检查甲状腺功能,测TSH 7.70 μIU/mL(参考值范围:0.27~4.20 μIU/mL),游离甲状腺素(free thyroxine,FT4)38.57 pmol/L(参考值范围:12.00~22.00 pmol/L),游离三碘甲腺原氨酸(free triiodothyronine,FT3)13.83 pmol/L(参考值范围:3.10~6.80 pmol/L),总三碘甲腺原氨酸(total triiodothyronine,TT3)4.62 nmol/L(参考值范围:1.30~3.10 nmol/L),总甲状腺素(total thyroxine,TT4)241.70 nmol/L(参考值范围:66.00~181.00 nmol/L),甲状腺过氧化物酶抗体(thyroid peroxidase antibody,TPOAb)、甲状腺球蛋白抗体(thyroglobulin antibody,TGAb)及促甲状腺激素受体抗体(thyroid stimulating hormone receptor antibody,TRAb)均正常。3岁时曾行甲状腺彩超检查提示双侧甲状腺形态正常,内部回声均匀,血流信号正常。定期随访期间无甲亢或甲减表现。
先证者6岁时再次因“甲状腺功能异常”就诊。查体:体温36.5 ℃,脉搏90次/min,呼吸18次/min,血压115/87 mmHg(1 mmHg=0.133 kPa),身高118 cm,体质量22 kg,精神正常,甲状腺无肿大、压痛,胸腹无异常。甲状腺结合球蛋白、性激素结合球蛋白、铁蛋白、肌酸激酶、总胆固醇、甘油三酯、低密度脂蛋白正常。听力测试示听力正常。甲状腺彩超示甲状腺左叶36 mm×14 mm×11 mm,右叶37 mm×14 mm×11 mm,峡部前后径2.1 mm,甲状腺形态大小正常,包膜完整,表面光滑,未见明显实质性团块,血流信号正常。垂体MRI平扫未见垂体瘤。左手腕关节X片提示符合正常骨龄表现。
5名家系成员包括其父亲、母亲、姑姑、爷爷、奶奶被纳入本研究。5名家系成员完善了甲状腺功能、甲状腺彩超检查。另选105例健康体检者(甲状腺功能正常)作为正常对照组。本研究已经通过医院伦理委员会批准(批准文号:2022k1)并符合《赫尔辛基宣言》。所有参与者均签署了知情同意书。
1.2 基因检测 采集所有受试者外周静脉血约3 mL于EDTA抗凝管中,提取基因组DNA。根据外显子侧翼的内含子序列设计引物。PCR扩增THRβ基因(MIM# 190160,GenBank NM_001128177.1)全部外显子。纯化PCR产物直接送至测序公司测序。疑似突变均经正、反测序证实。对于疑似突变,应用Mutation Taster、SIFT、Polyphen-2、PROVEAN生物信息学软件对其进行功能预测,分析其致病性。引物设计、合成及测序由北京中美泰和生物技术有限公司完成。
1.3 随访方法 先证者每2~6个月随访1次,检查生长发育、智力发育、甲状腺功能、甲状腺彩超等。先证者父亲每年随访1次,完善甲状腺功能及甲状腺彩超检查,评估有无异常临床表现。
2 结果
2.1 初诊结果 5个家系成员中,先证者父亲甲状腺功能表现为正常的TSH和升高的甲状腺激素,符合RTH(见表1)。先证者父亲甲状腺超声提示甲状腺左叶低回声结节,甲状腺ECT检查未见明显异常。该家系中其他成员甲状腺功能、彩超检查均正常。
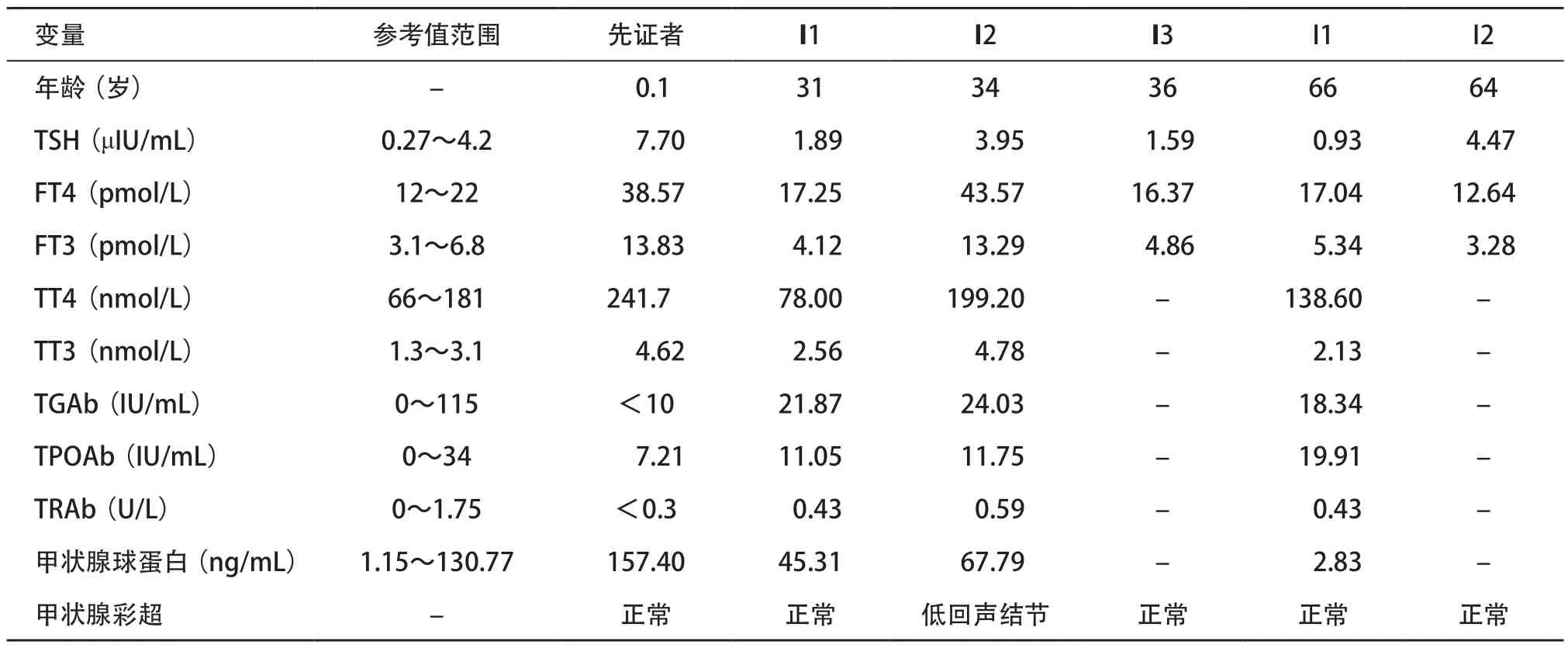
表1 诊断时先证者及家系成员甲状腺相关检查结果
2.2 基因检测结果 基因测序结果显示,先证者及其父亲携带THRβ基因c.728G>A(p.Arg243Gln,p.R243Q)杂合变异,遗传方式符合常染色体显性遗传(见图1)。THRβ基因c.728G>A突变导致第7外显子243位精氨酸被谷氨酰胺取代。THRβ基因突变型与野生型DNA序列如图2所示。该家系中其他成员和105例健康对照组未发现相同突变位点。

图1 RTH患者的家系图(箭头所指为先证者)
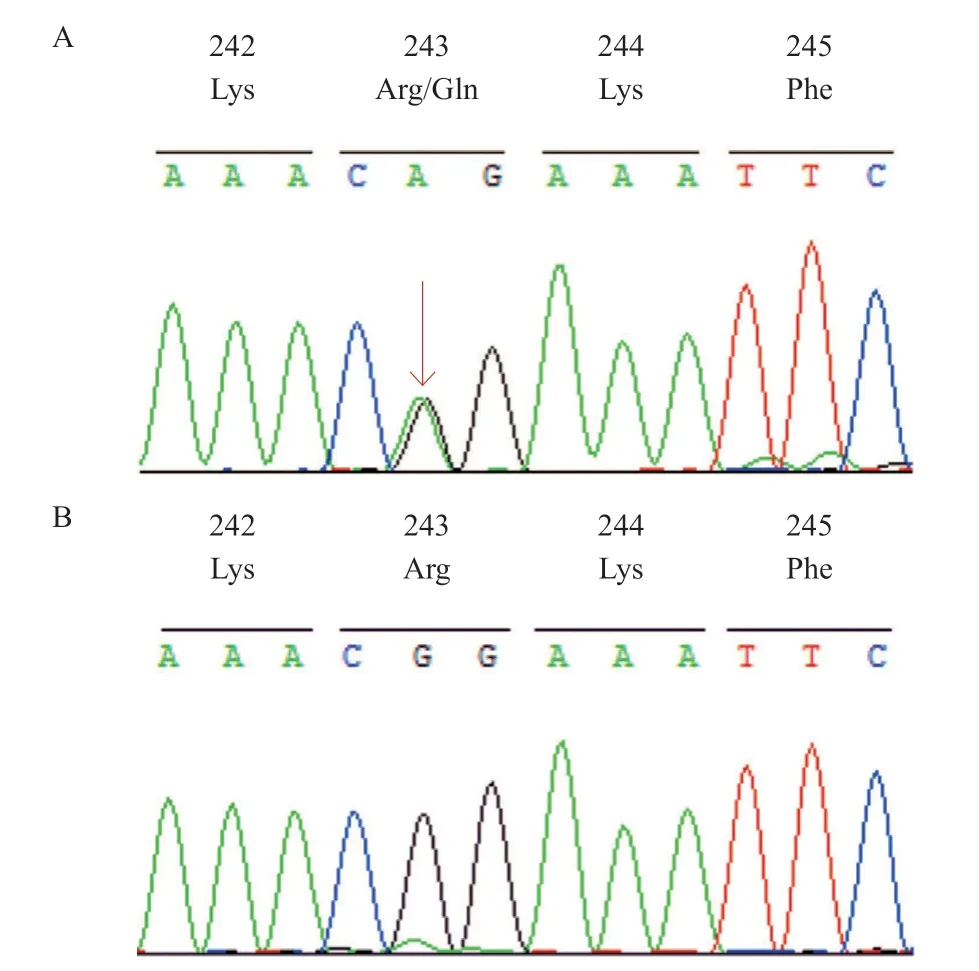
图2 THR β基因第7外显子测序结果(局部)
2.3 随访结果 6年随访期间,先证者甲状腺功能如图3所示,TSH、FT3、FT4始终高于正常参考范围,符合RTH血清学特点。生长发育如图4所示。学习成绩中等偏上,5岁时曾行中文版韦氏幼儿智力量表(第4版)测试,结果显示为中上水平。6岁时再次行中文版韦氏幼儿智力量表(第2版)测试,结果显示为中上水平。先证者父亲每年定期复查甲状腺功能,均符合RTH血清学特点,多次行甲状腺彩超示甲状腺形态大小正常,左侧叶低回声结节,直径小于1 cm。先证者及父亲均无怕热多汗、心慌手抖、多食易饥等甲亢症状或多动症,以及怕冷乏力、腹胀便秘、记忆力减退等甲减症状。

图3 先证者甲状腺功能6年随访结果(灰色条段为参考值范围)
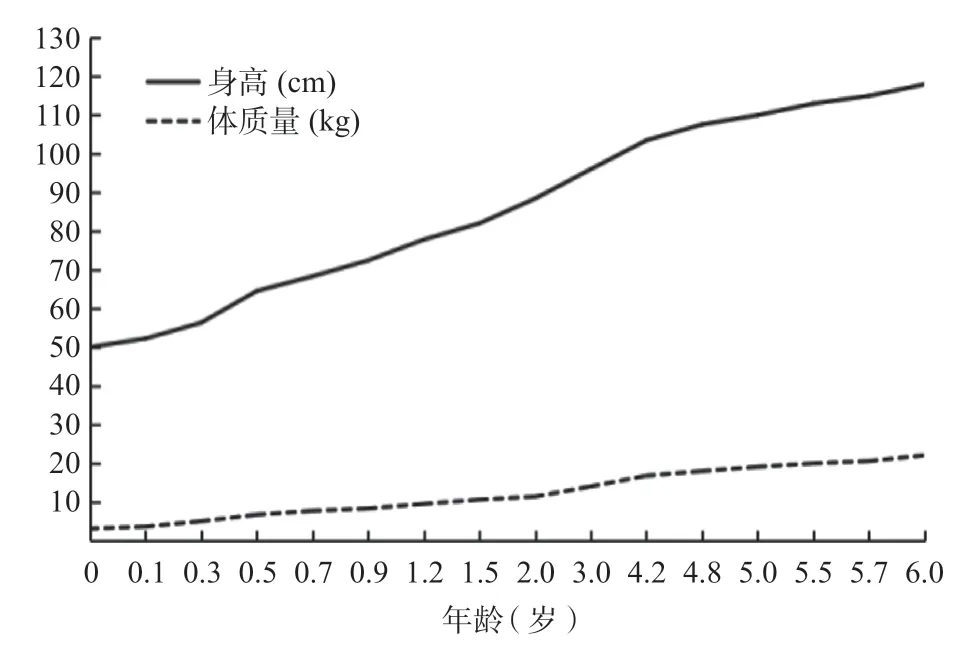
图4 随访期间先证者生长发育图
3 讨论
RTH是一种罕见遗传病,可表现为孤立的甲功异常、甲亢或甲减,临床中容易误诊、漏诊。目前国内报道该病大部分患者被误诊为甲亢而予抗甲亢药物或131I、手术等不恰当的治疗方案[9],因此有必要对罕见病例进行报道,提高临床医师对该病的认识,降低漏诊、误诊率。
通过新生儿筛查而确诊RTH 的病例较为少见[10]。本研究中,先证者是在新生儿筛查时发现异常甲状腺功能,提示RTH,无异常临床表现。先证者父亲因出生时宿迁地区尚未开展新生儿筛查工作,且无甲亢或甲减临床表现而延误了RTH诊断[11]。进一步行基因测序结果显示,先证者及其父亲均携带THRβ基因c.728G>A(p.R243Q)杂合突变,遗传方式符合常染色体显性遗传。
THRβ基因c.728G>A突变极为罕见,仅见于极少数RTH患者,首次由国外学者[7,12-14]报道,目前国内仅报道1例患者。THR β属于核受体家族中的一员,是一种可被T3激活的转录因子,进而影响靶基因表达。THR β包含转录激活域、DNA结合域、铰链区、配体结构域。大部分的THRβ基因突变可不同程度直接影响T3结合力而导致RTH[1]。与其他突变不同的是,c.728G>A突变位于第7外显子编码的铰链区,靠近配体结构域的N末端。前期功能学研究显示,c.728G>A突变可影响THR β配体结构域N末端的流动性和灵活性而降低配体结构域的整体稳定性,从而显著影响辅助抑制因子的释放并轻度降低T3结合力[15-18]。因此,甲状腺激素水平的升高可能是对辅助抑制因子释放缺陷和T3结合力降低的代偿。正常或升高的TSH是甲状腺激素负反馈缺失的表现。
根据各组织对甲状腺激素的抵抗程度不同,RTH可分为全身抵抗型、选择性垂体抵抗型、选择性外周抵抗型。其中,全身抵抗型比较常见,大部分患者表现为正常代谢状态而无异常临床表现,少部分表现为甲减[19]。本研究中,2例携带R243Q杂合突变的RTH患者无甲状腺肿、甲亢或甲减的临床表现,垂体MRI未见异常,考虑为全身抵抗型。ONIGATA等[12]报道携带相同突变位点的家系中,先证者无异常临床表现,先证者母亲表现为甲状腺肿和甲亢表现。而国内学者报道的携带R243Q杂合突变的先证者仅表现为甲状腺肿,诊断为RTH(全身抵抗型)[7]。携带相同突变位点患者的临床表型不同以及同一家系中携带相同突变位点患者的临床表型不同提示了RTH临床表型的异质性。因此仍需长期随访2例RTH患者,及早发现有无甲状腺肿或其他异常临床表现。
随访过程中,先证者父亲甲状腺彩超提示甲状腺左叶低回声结节,直径小于1 cm,暂无其他恶性征象。甲状腺结节低回声的超声特征与恶性肿瘤风险增加有关,中度或明显低回声结节的恶性风险高于轻度低回声结节[20]。近年来国内外学者先后报道16例RTH合并甲状腺乳头状癌(papillary thyroid carcinoma,PTC)的患者,大部分患者因甲状腺结节或甲状腺肿就诊,无明显甲亢症状。他们接受了甲状腺全切/次全切和(或)颈部淋巴结清扫,病理证实PTC,术后予L-T4替代治疗以抑制TSH低水平和维持正常水平的甲状腺激素,需要剂量较大,最高可达500 μg/d[21-22]。RTH是否增加PTC以及侵袭性风险尚不清楚。部分学者推测,长期高水平TSH对甲状腺的持续性刺激可能与甲状腺结节和甲状腺癌形成有关[23]。同时,就目前已报道的RTH合并PTC病例中,大部分患者携带THRβ基因突变。THRβ基因突变在肝癌、肾癌、乳腺癌、垂体瘤、结肠癌等中被报道过[24]。THRβ基因突变在PTC形成中可能发挥一定的致癌作用。RAMOS-PROL等[25]曾报道1例RTH合并PTC的9岁女童,该患者携带THRβ基因R243W突变。因此,本研究中2例携带THRβ基因R243Q突变患者是否发生相关肿瘤,需长期随访甲状腺彩超,必要时行超声引导下细针穿刺活检术。
目前对于RTH尚无根治方法。RTH患者的治疗应该以维持正常代谢状态为目标。先证者及其父亲均无甲亢或甲减的临床表现,考虑与升高的甲状腺激素水平弥补了机体对甲状腺激素的不敏感性有关,暂未加用药物治疗。
综上所述,本研究的先证者及其父亲携带THR β基因c.728G>A杂合突变,是RTH发病的遗传学病因,仍需长期随访2例患者临床表型。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
中国典型病例大全(2022年7期)2022-04-22
自我保健(2021年3期)2021-05-12
郑州大学学报(医学版)(2019年3期)2019-06-03
传染病信息(2019年2期)2019-05-17
中国卫生标准管理(2015年25期)2016-01-14
广东海洋大学学报(2015年4期)2016-01-13
听力学及言语疾病杂志(2015年5期)2015-12-24
首都医科大学学报(2015年4期)2015-12-16
重庆医学(2015年12期)2015-03-05
