
基于UPLC-QTOF-MS/MS分析的芪葵颗粒化学成分系统筛选与识别
2023-02-09 12:34李长印陆明霞廖健城黄莉吉居文政邹建东
南京中医药大学学报 2023年1期
李长印,陆明霞,廖健城,黄莉吉,居文政,邹建东
(1.南京中医药大学附属医院,江苏省中医院临床药理实验室,江苏 南京 210029;2.南京中医药大学附属医院,江苏省中医院内分泌科,江苏 南京 210029)
芪葵颗粒为江苏省中医院广泛应用的治疗早中期糖尿病肾病的院内制剂,全方由黄芪、制首乌、黄蜀葵花3味中药依据“益气养阴、清利活血”的早中期2型糖尿病肾病治疗原则组方而成。方中黄芪甘温,补中益气,升阳止渴,利水退肿;制首乌补血益精,滋补肝肾,且微温而不燥热,补虚而不滋腻,二药配用,气阴兼顾,益气养阴,养血理血;配以黄蜀葵花,利水活血,通淋消肿,补泻同施,标本兼顾。芪葵颗粒临床疗效确切[1-2],且与当前治疗糖尿病肾病的临床常用西药相比,具有改善患者整体症状和长期服用安全有效等优势,因而具有巨大的开发应用潜力。目前,芪葵颗粒的相关研究多集中于临床疗效[1-2]及药理机制[3-4]方面,缺乏对其化学成分系统全面的分析和鉴定。超高效液相色谱-四级杆飞行时间质谱联用技术(UPLC-QTOF-MS/MS)兼具UPLC的高效分离能力以及TOFMS的高灵敏度、高分辨率、高质量精度和扫描范围广等特点,现已广泛应用于中药复杂化学成分的系统定性表征[5-6]。近年来,本课题组以此联用分析技术为基础,先后对芪葵颗粒组方的单味药材黄芪[7-9]和黄蜀葵花[10]进行了较为系统的化学成分定性分析。在此基础上,本研究首先采用UPLC-QTOF-MS/MS方法对芪葵颗粒复方进行分析检测,进而借助PeakView软件,综合运用靶向和非靶向筛选策略,全面筛选并鉴定复方中含有的主要化学成分,以明确复方中化学成分的存在形式,为明确、优化复方制剂质量控制标准以及深入研究复方制剂的药效物质基础提供依据。
1 材料
1.1 实验仪器
Agilent 1290 Infinity超高效液相色谱系统(美国Agilent公司),配备有柱温箱G1316C、自动进样器G4226A,自动进样器控温模块G1330B和二元泵G4220A;Triple TOFTM5600高分辨质谱仪,配备Analyst TF 1.6操作系统及PeakView 2.2数据处理软件(美国AB Sciex公司);CDS质谱自动校准系统;SorvallTMLegendTMMicro 17R微量离心机(Thermo Scientific公司);WH2微型旋涡混合仪(上海沪西分析仪器厂);CPA225D电子天平(德国Sartorius公司);Millipore Milli-Q Advantage A10超纯水系统(Milli-Q公司)。
1.2 试剂与药物
甲醇、乙腈(HPLC级,德国Merck公司);甲酸、甲酸铵(质谱级,瑞士Fluka-Sigma-Aldrich公司);TOF-MS正、负离子调谐液(美国AB Sciex公司,批号:4460131、4460134)。
芪葵颗粒由江苏省中医院制剂部制备,每袋10 g,批号:1607001;20个芪葵颗粒相关化学成分对照品的名称、生产厂家和批号等信息详见表1。
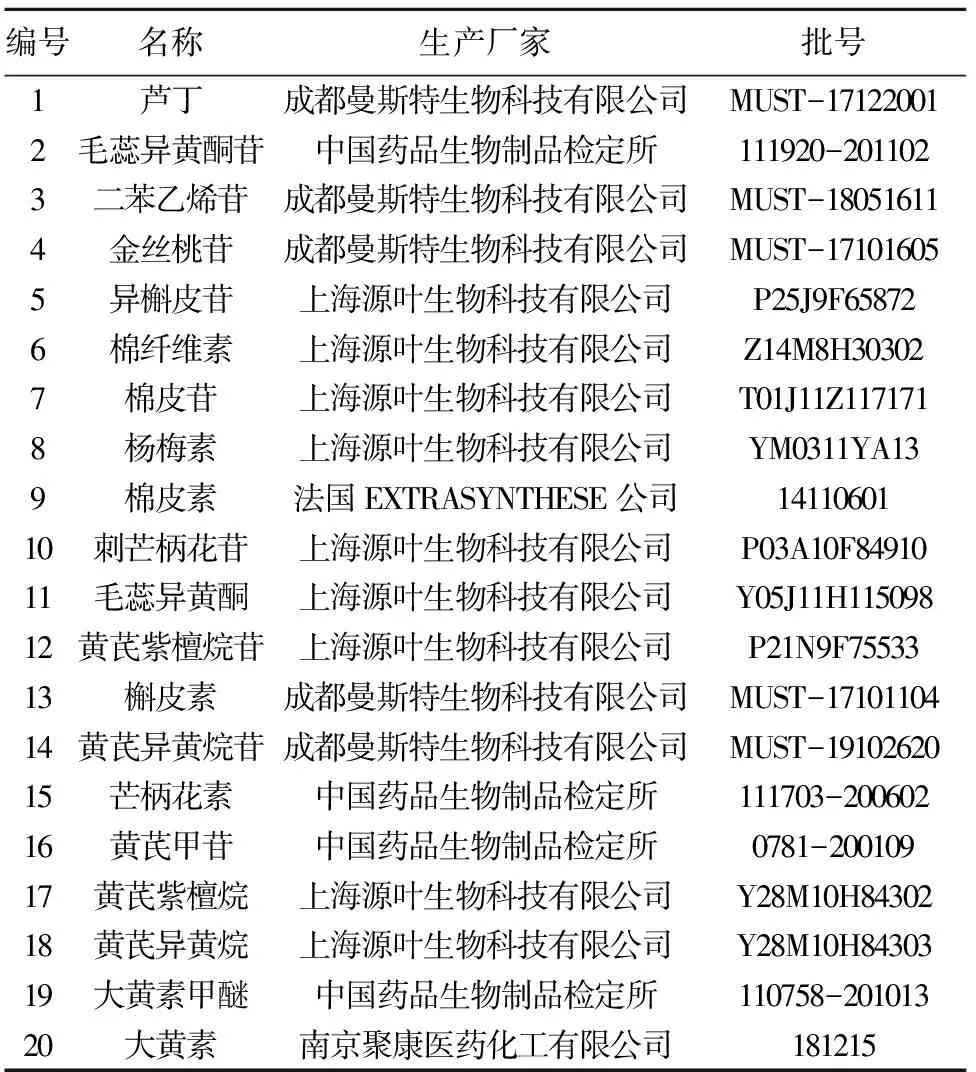
表1 芪葵颗粒20个主要化学成分对照品信息Table 1 General information of 20 authentic standards for major constituents in Qikui Granules
2 方法
2.1 样品制备
精密称取芪葵颗粒105.8 mg,加入超纯水或纯甲醇5 mL,超声提取(50 kHz,300 W)20 min,涡旋30 s混匀后,立即精密吸取1 mL于1.5 mL离心管中,4 ℃ 12 000×g离心5 min,取上清液进行UPLC-QTOF-MS/MS分析。
精密称取20个对照品适量,以甲醇稀释至适当浓度,并等量混合制备混标溶液,取混标及单标溶液进行UPLC-QTOF-MS/MS分析,对定性鉴定结果进行比对确认。
2.2 色谱条件
采用Agilent 1290 Infinity超高效液相色谱系统进行色谱分离。色谱柱:Agilent Poroshell 120 SB-C18色谱柱(3.0 mm×100 mm,2.7 m);柱温:45 ℃;流速:0.4 mL·min-1;流动相:A相为0.1%甲酸的水溶液,质谱负离子检测模式下B相为含有10 mmol·L-1甲酸铵的乙腈/水(95∶5,v/v),质谱正离子检测模式下B相为含有10 mmol·L-1甲酸铵和0.125%甲酸的乙腈/水(95∶5,v/v);采用梯度洗脱分离:0~6 min,90%A;6~12 min:90%~80%A;12~18 min:80%~65%A;18~23 min:65%~50%A;23~28 min,50%~25%A;28~32 min:25%A;32~35 min:25%~90%A;35~45 min,90%A。进样体积:正离子模式下为3 μL,负离子模式下为6 μL;自动进样器温度4 ℃。
2.3 质谱条件
采用AB SCIEX Triple TOFTM5600质谱仪进行检测。采用电喷雾离子源(ESI)在正、负2种离子模式下进行采集。TOFMS扫描模式参数设置如下:雾化气(GS1):60 psi;辅助加热气(GS2):60 psi;气帘气(CUR):35 psi;离子化温度(TEM):500 ℃;正、负离子模式下喷雾电压(ISVF)分别为5 500 V和4 500 V;分子量扫描范围:m/z50~1 000,累积时间:0.15 s;去簇电压(DP):80 V;碰撞能量(CE):10 eV。采用动态背景扣除(DBS)和触发信息关联采集模式(IDA)采集数据。主要的IDA转换标准如下:信号强度大于50 cps,4 g·mL-1以内排除同位素,分子量误差50 mg·mL-1,每个循环最多监测12个候选离子。子离子扫描模式的主要参数如下:分子量扫描范围m/z25~1 000,累积时间0.035 s;CE:(40±20)eV;其他参数同TOFMS扫描模式。采用AB公司的调谐液传递系统(CDS)对分子量准确度进行自动校准。数据采集过程由Analyst TF 1.6进行控制。
2.4 数据分析
2.4.1 化合物筛选 采用目标化合物靶向筛选和主要化学成分非靶向筛选相结合的方式研究芪葵颗粒复方中的化学成分。
目标化合物的靶向筛选流程如下:①依据前期黄芪[7-9,11-12]、黄蜀葵花[10]和何首乌[13-18]三味药材化学成分研究相关文献报道,建立芪葵颗粒化学成分库;②在芪葵颗粒提取液的负离子模式下UPLC-QTOF-MS/MS总离子流色谱图(TIC)中,利用PeakView软件的提取离子流色谱图(XIC)功能,获取芪葵颗粒数据库各化合物目标离子(即[M-H]-、[M+HNO3-H]-和[M+HCOOH-H]-三种准分子离子)的XIC图;③考察各XIC图谱中出现的每个色谱峰所对应的TOFMS质谱图,依据TOFMS提供的精确实测分子质量(与库化合物的精确理论分子质量差值应<5×10-6)确证目标离子的存在并记录其准确保留时间;④在相应保留时间的TOFMS质谱图中,结合质量亏损规律[7-8,10]筛选质荷比(m/z)大于库化合物[M-H]-离子的可能相关离子,以排除碎片离子的干扰,并依据各离子精确分子质量间的相互关联,筛选潜在目标化合物的各种可能类型的准分子离子(如[M-H]-、[M+HNO3-H]-、[M+HCOOH-H]-、[M+HCl-H]-、[2M-H]-等);⑤依据上述准分子离子的类型和精确分子质量,结合芪葵颗粒数据库中化合物的元素组成,推断筛选所得化合物的化学元素组成(即分子式);⑥在正离子模式下,采用PeakView软件的XIC功能,提取各分子式对应不同类型准分子离子(如[M+H]+、[M+Na]+、[M+NH4]+、[M+K]+、[2M+H]+等)的XIC图,在与负离子模式相对应的保留时间处考察其TOFMS质谱图,确证对应化合物上述准分子离子的存在;⑦结合正、负离子筛选结果,以至少2个准分子离子的出现为标准确认筛选化合物的存在,锁定目标化合物。
为了全面系统表征芪葵颗粒复方的化学成分,弥补基于已知化合物库筛选的不足,同时进行复方主要化学成分的非靶向筛选。采用PeakView非靶标峰寻找(Non-Targeted peak finding)功能,设定强度阈值为9 000 cps,全面提取负离子模式下芪葵颗粒中的主要化学成分离子,以部分确证上述靶向筛选结果,并按照上述流程③④⑦尝试对丰度较大的未知化学成分进行锁定。
2.4.2 化合物结构鉴定和归属 锁定目标化合物后,依据上述芪葵颗粒数据库及相关文献报道,初步确定化合物名称和结构,并通过对照品比对,准确鉴定部分化合物;同时结合文献报道对目标化合物的二级质谱碎片离子进行解析,并与AB Sciex中药高分辨质谱数据库(数据库匹配得分>95)搜索结果、文献报道或对照品的二级信息进行比对,确证或合理归属目标化合物的具体化学结构。
2.4.3 鉴定结果梳理 根据化学成分结构的相似性和关联,结合可能发生的转化反应,对鉴定出的各化合物间的关联性和相互间代谢及转化关系进行梳理和总结,以表征芪葵颗粒复方化学成分的多样性和相互关联性。
3 结果
3.1 化合物筛选
采用UPLC-QTOF-MS/MS在“2.2”色谱和“2.3”质谱条件下对芪葵颗粒水提液分别进行进样分析,获得正、负离子模式下的TIC图,如图1a~b所示。按照“2.4.1”化合物筛选流程,最终从芪葵颗粒水提液中筛选锁定了89个目标化合物。各化合物正、负离子模式下的叠加XIC图如图1c~d所示,大部分化合物在现有色谱条件下可以得到比较好的分离;而对于少数不能很好分离的化合物,则可以通过高分辨质谱的区分能力和丰富的准分子离子类型加以区分和确认。
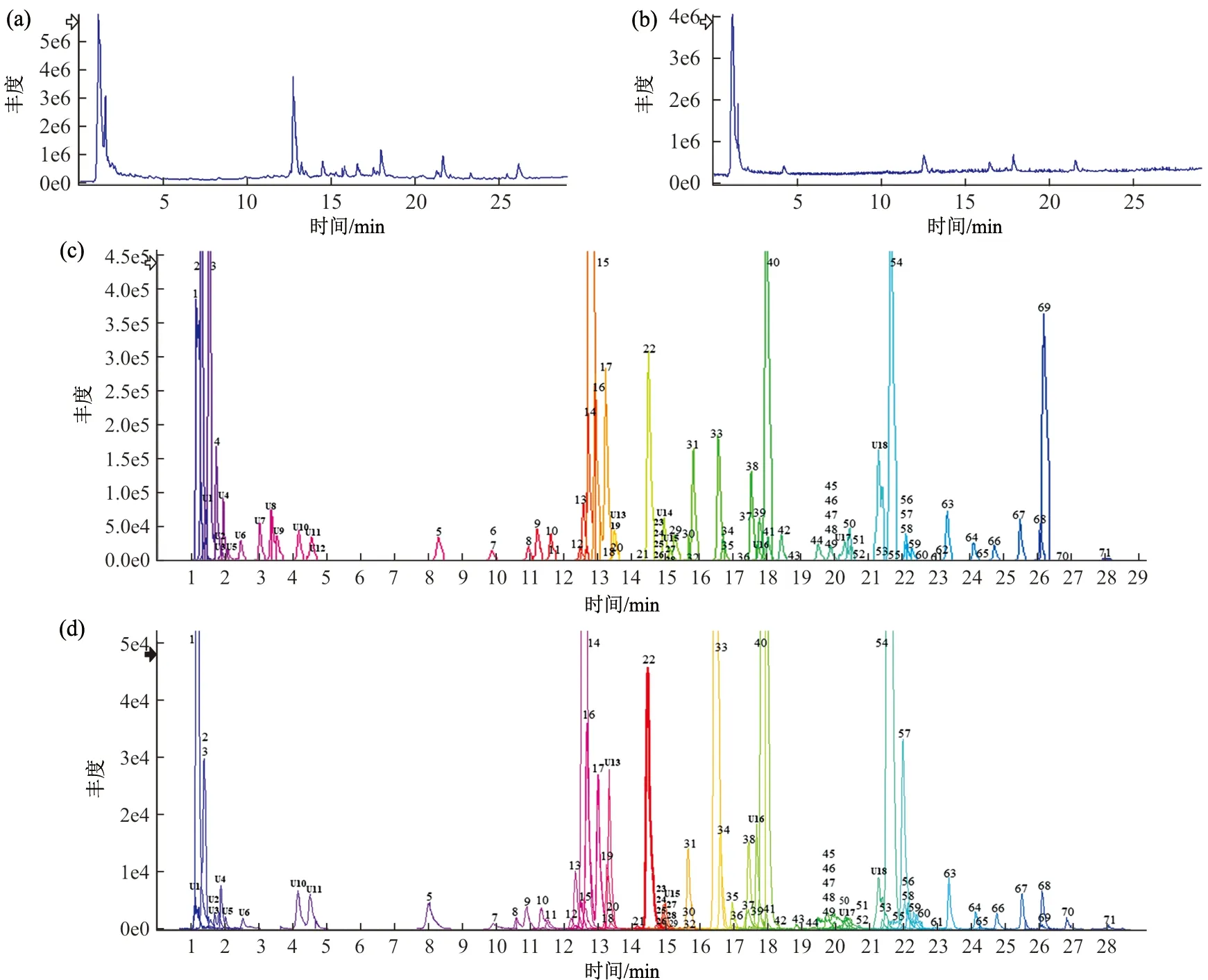
注:a.负离子模式下TIC图;b.正离子模式下TIC图;c.负离子模式下XIC图;d.正离子模式下XIC图;1~71.已鉴定化学成分LC1~LC71;U1~U18.未鉴定化学成分图1 芪葵颗粒水提液的UPLC-QTOF-MS色谱图Fig.1 The UPLC-QTOF-MS chromatograms from the aqueous extract of Qikui Granules
3.2 化学成分鉴定
对89个化合物中的71个化合物实现了合理的结构鉴定或归属。表2汇总了71个已鉴定化合物的保留时间(tR)、分子式、准分子离子、峰强度、子离子、名称、来源药材等详细信息。其中19个化合物通过对照品比对进行了确认;18个化合物的一、二级质谱信息与AB Sciex中药高分辨质谱数据库匹配良好(匹配得分均大于95,满分为100)。现将主要鉴定过程简述如下。
3.2.1 黄酮类 作为黄芪和黄蜀葵花2味药材的主要化学成分,黄酮类化合物应为芪葵颗粒复方制剂的主要化学和药效成分。在本研究筛选鉴定的71个化合物中,有多达45个化合物被归属为黄酮类,其中16个可进一步通过对照品比对准确鉴定。45个黄酮类成分按药材来源可大体划分为黄芪黄酮和黄葵黄酮两大类。
3.2.1.1 黄芪黄酮类 共有31个化合物归属为黄芪黄酮类,其中8个化合物经对照品比对准确鉴定。
如表2所示,多个加合离子和精确分子质量信息提示,LC54的分子式可能为C16H12O4,对应复方中黄芪主要成分之一芒柄花素,其特征性碎片离子m/z267、252、223、208、195、167、135、132等的出现,以及对照品的比对结果表明,LC54为芒柄花素。类似地,LC40、LC60、LC57分别被准确鉴定为毛蕊异黄酮、黄芪异黄烷和黄芪紫檀烷。LC33、LC14、LC41和LC38可通过标准品比对分别被准确鉴定为上述4个苷元的葡萄糖苷类化合物芒柄花苷、毛蕊异黄酮苷、黄芪异黄烷苷和黄芪紫檀烷苷,它们均可特征性地丢失1分子葡萄糖(162)生成对应苷元离子,并进一步裂解产生苷元特征子离子。上述4个苷元LC54、LC40、LC60、LC57为黄芪黄酮类化合物的主要结构母核,其结构中羟基、甲基、甲氧基、糖基等取代基团的位置信息可为复方中其他相关化合物的结构归属提供合理依据。各苷元的裂解途径详见前期文献报道[9,12],裂解产生的特征子离子(表2)对于解析其结构类似物和相应糖苷类化合物具有重要指导意义。
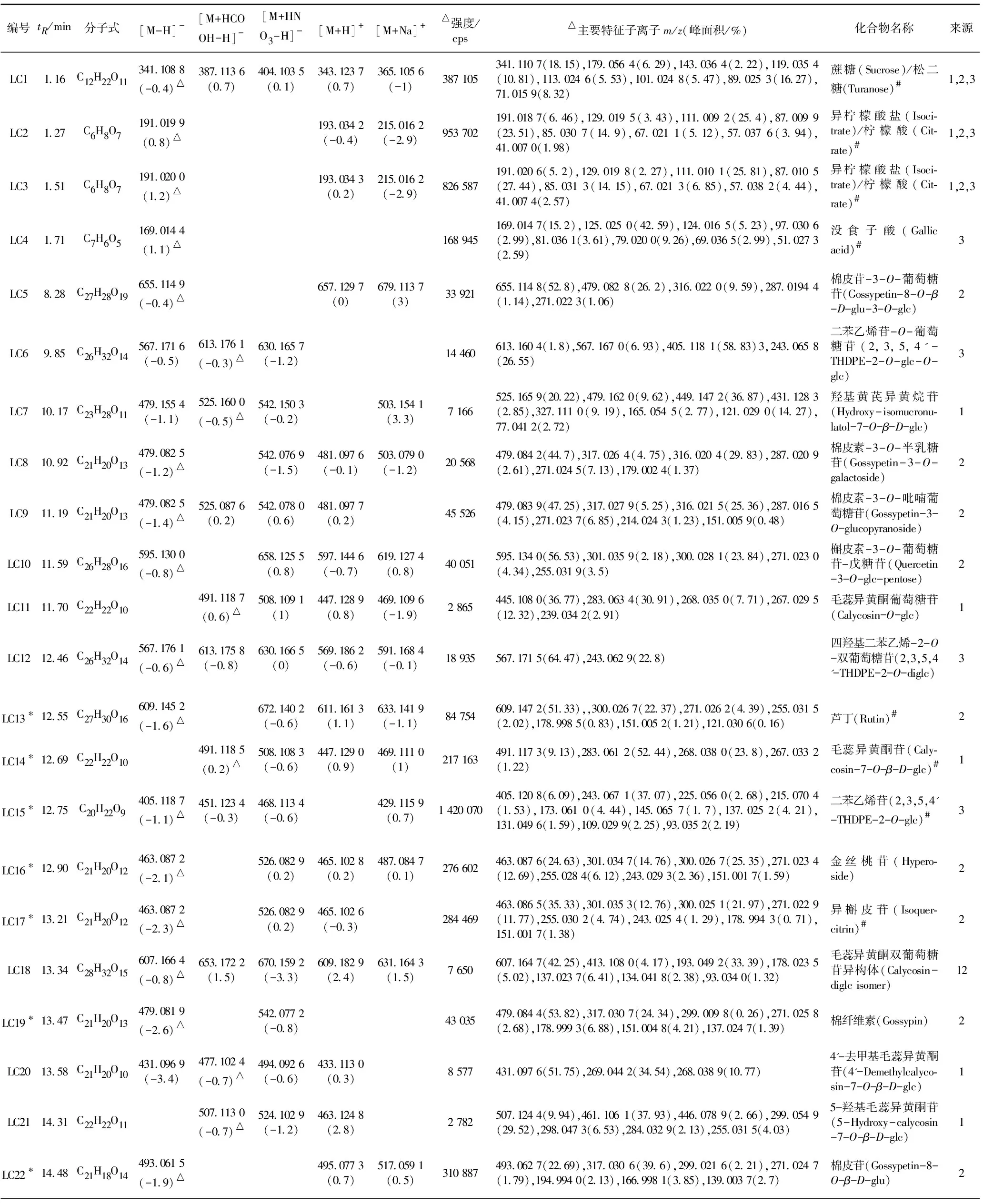
表2 芪葵颗粒中鉴定出的71个化合物的详细信息Table 2 The detailed information of 71 identified compounds in Qikui Granules
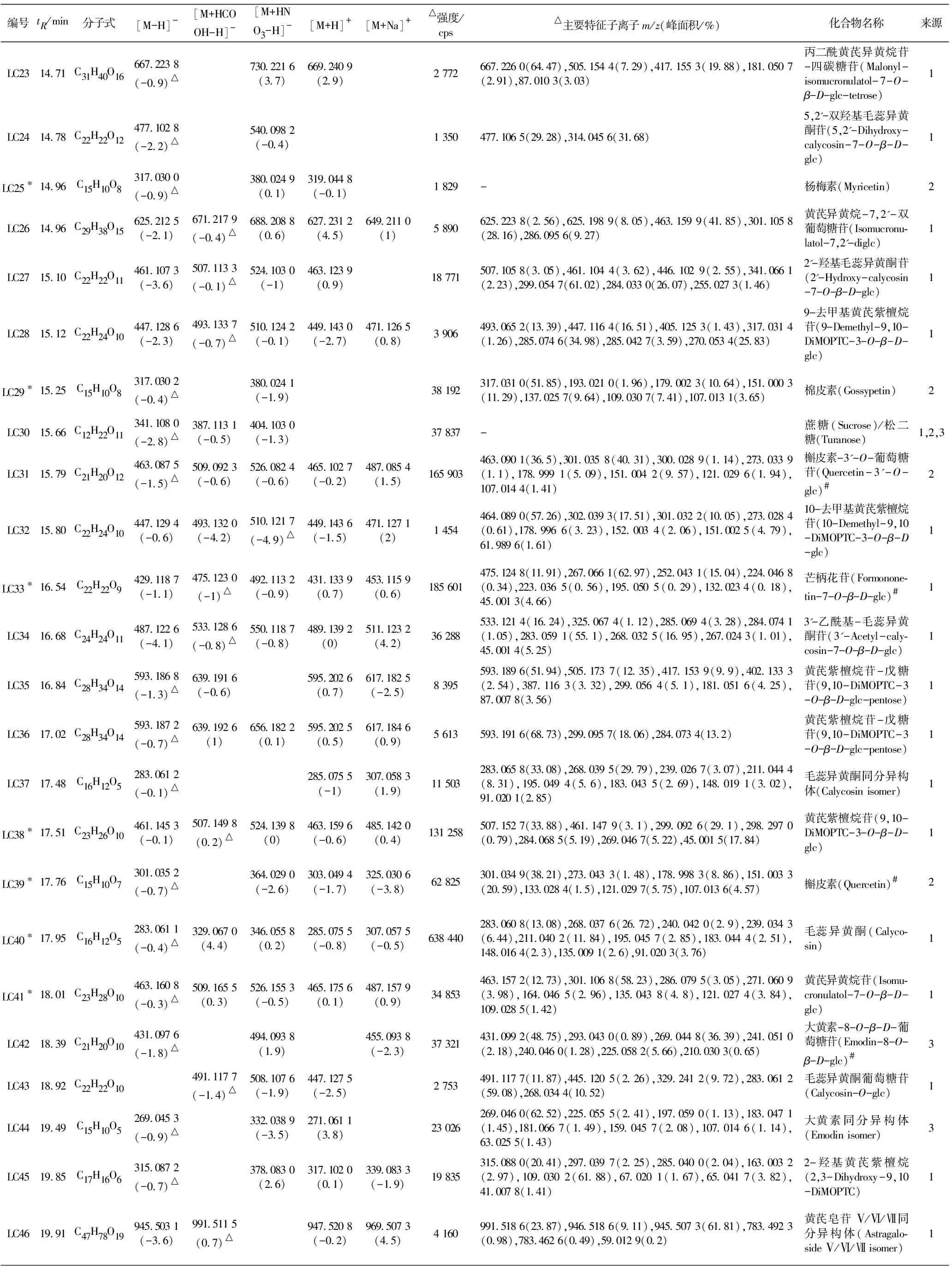
(续表一)
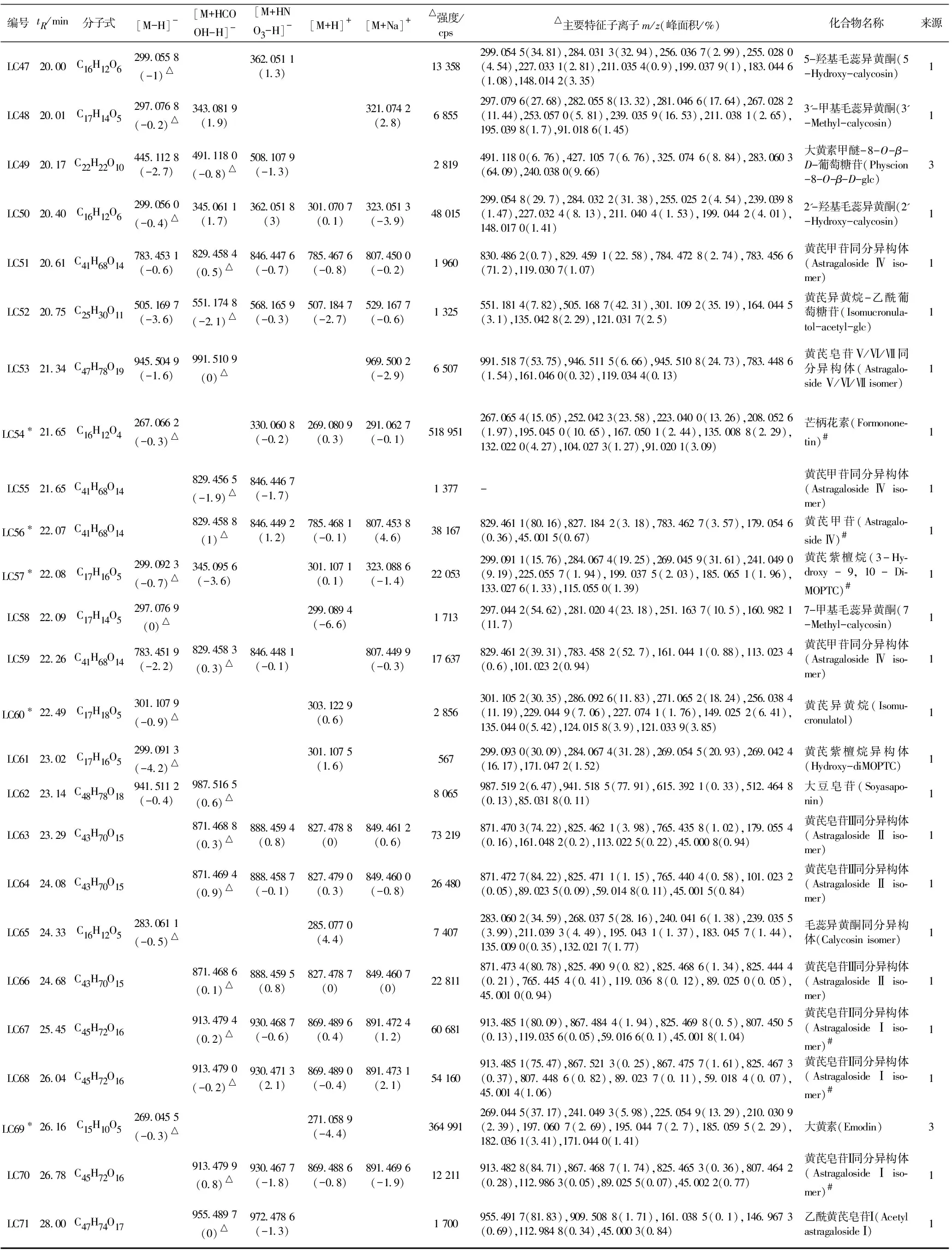
(续表二)
需要注意的是,复方中何首乌药材中可能含有的蒽醌苷元大黄素甲醚[14,16-17]与毛蕊异黄酮互为同分异构体。对照品进样分析结果显示,由于化合物极性不同,两者保留时间相差很远,分别为29.25 min和17.96 min,且两者产生的主要特征子离子完全不同。毛蕊异黄酮的主要特征子离子为m/z268、239、211、183、148、135、91等(图2a),而大黄素甲醚的主要特征子离子为m/z240、212、184等(图2b),这些离子的出现可用于区别鉴定与两者结构类似的相关化合物。根据上述毛蕊异黄酮主要特征子离子的出现,初步鉴定出6个结构类似物。其中LC37和LC65与毛蕊异黄酮分子式相同,且同样可产生主要特征子离子m/z268、239、211、183,初步鉴定其为毛蕊异黄酮同分异构体。LC47和LC50分子式比毛蕊异黄酮多一个O,它们既可产生m/z239、211、183、148等毛蕊异黄酮特征子离子,又可产生比其多16(羟基)的299、284、255、227、199等系列特征碎片离子,据此将其归属为羟基毛蕊异黄酮;参考黄芪黄酮苷元LC60、LC57和黄葵黄酮苷元LC39的苯环羟基取代位置,初步推断羟基化位点为2'位或5位;综合结构类似性和丰度信息,将LC47和LC50分别初步归属为5-羟基毛蕊异黄酮和2'-羟基毛蕊异黄酮。LC48和LC58为一组同分异构体,其特征性母离子m/z297[M-H]-提示其分子式为C17H14O5,比毛蕊异黄酮(LC40)的分子式C16H12O5多CH2,提示其可能为甲基化的毛蕊异黄酮,甲基化位点可能为毛蕊异黄酮的3'位或7位羟基;考虑到黄芪异黄烷(LC60)的化学结构为3'位甲氧基取代,且LC48的丰度(6 855 cps)远高于LC58(1 713 cps),LC48和LC58分别被初步鉴定为3'-甲基毛蕊异黄酮和7-甲基毛蕊异黄酮。LC11、LC14和LC43为一组毛蕊异黄酮苷的同分异构体,他们均可通过特征性地丢失1分子葡萄糖(162)生成苷元离子m/z283以及高丰度的毛蕊异黄酮特征子离子m/z268,其中LC14可通过标准品的保留时间和一、二级质谱图(图2c)比对被准确鉴定为毛蕊异黄酮苷,这也在一定程度上验证了依据上述苷元特征子离子归属类似化合物结构的可靠性。元素组成信息提示LC21和LC27为羟基化的LC14,L24为双羟基化的LC14;对应于LC47和LC50,将LC21、LC27和L24分别初步归属为5-羟基毛蕊异黄酮苷、2'-羟基毛蕊异黄酮苷和5,2'-双羟基毛蕊异黄酮苷,其二级碎片离子中作为基峰离子存在的苷元离子m/z299、314可为上述结构归属提供进一步的佐证。类似地,LC34的分子式提示为乙酰化的LC14,苷元子离子m/z325表明,乙酰化不是发生在葡萄糖残基上,而是3'位羟基上,据此鉴定其为3'-乙酰基毛蕊异黄酮苷。LC20的元素组成提示其为去甲基化的LC14,由于LC14结构中仅4'位有甲氧基,据此推断其为4'-去甲基毛蕊异黄酮苷,苷元4'-去甲基毛蕊异黄酮对应的碎片离子m/z269可为此推断提供确证依据。
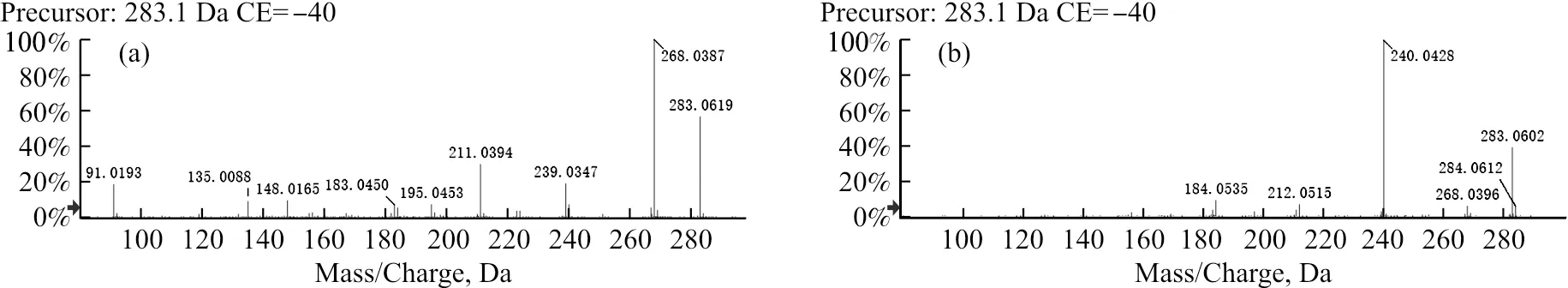

注:a和b源自对照品溶液;c和d源自芪葵颗粒水提液,对应编号为LC14和LC49的目标化合物。图2 毛蕊异黄酮(a)、大黄素甲醚(b)、毛蕊异黄酮苷(c)和大黄素甲醚-8-O-β-D-葡萄糖苷(d)的二级子离子图谱Fig.2 Product ion mass spectra of calycosin (a), physcion (b), calycosin-7-O-β-D-glucoside (c) and physcion-8-O-β-D-glucoside (d)
元素组成信息提示LC7(C23H28O11)、LC23(C31H40O16)、LC26(C29H38O15)和LC52(C25H30O11)的结构与LC41(C23H28O10)相关。LC7可能为羟基化LC41,但没有进一步的子离子提供支持,仅将其初步鉴定为羟基黄芪异黄烷苷,并在图3中给出了一种可能的化学结构;LC52为乙酰化LC41,可产生对应黄芪异黄烷的基峰子离子m/z301,且未出现乙酰化黄芪异黄烷对应的子离子,表明黄芪异黄烷即为其苷元,乙酰化葡萄糖为其糖苷,据此将其初步鉴定黄芪异黄烷-乙酰葡萄糖苷;类似地,LC26可先后丢失1个葡萄糖残基(162)产生高丰度子离子m/z463、301,分别对应LC41和黄芪异黄烷(LC60),且LC41对应的子离子丰度明显高于黄芪异黄烷,提示2个葡萄糖残基先后丢失比共同丢失的几率更大,推测2个葡萄糖残基可能位于不同的羟基取代位点,据此可将其初步鉴定为黄芪异黄烷-7,2'-双葡萄糖苷;LC23由于丰度较小,仅可见微量的丢失1分子葡萄糖(162)的碎片离子m/z505,结合文献报道[9,11],将其初步鉴定为丙二酰黄芪异黄烷苷-四碳糖苷。
LC61为LC57的同分异构体,且可通过特征性先后丢失O和CH2产生LC57的特征子离子m/z284、269等,表明其结构中同样含有1个羟基和1个甲基,据此初步将其归属为黄芪紫檀烷异构体。LC45的分子式为C17H16O6,比苷元LC57多一个O,且其可产生[M-H2O]-特征子离子m/z297,提示其结构中的2个羟基可能位于邻位,据此初步推断其为2-羟基黄芪紫檀烷。LC35和LC36的分子式均为C28H34O14,比LC38(C23H26O10)多1个戊糖残基(C5H8O4),且可产生共同的子离子m/z299,对应苷元LC57;由于LC38结构中仅有葡萄糖残基上可能发生戊糖取代,初步推断LC35和LC36为一组黄芪紫檀烷苷-戊糖苷的同分异构体,戊糖类型及取代位点待定,仅在图3中给出一种可能结构。LC28和LC32的分子式为C22H24O10,比LC38少CH2,提示两者为去甲基化的LC38,这与LC38的苷元中含有2个甲氧基相吻合,但现阶段无法确定两者的对应关系,因此,暂将LC28和LC32归属为9-去甲基黄芪紫檀烷苷或10-去甲基黄芪紫檀烷苷。
3.2.1.2 黄葵黄酮类 共有13个化合物归属为黄葵黄酮类,其中8个化合物经对照品比对准确鉴定。
通过解析一级、二级质谱信息,并与对照品的保留时间和相关质谱信息进行比对,LC39、LC25、LC29被准确鉴定为槲皮素、杨梅素和棉皮素。如表2所示,其苷元特征子离子可为鉴定其结构类似物和相应糖苷类化合物提供重要参考。LC16和LC17被准确鉴定为槲皮素的糖苷金丝桃苷和异槲皮苷,参考色谱保留行为与前期研究的一致性[19],其同分异构体LC31被暂时鉴定为黄蜀葵花的另一个主要黄酮类成分槲皮素-3'-O-葡萄糖苷。LC13、LC19和LC22分别被准确鉴定为芦丁、棉纤维素和棉皮苷。如图2所示,上述经标准品比对准确鉴定的5个糖苷类成分LC16、LC17、LC13、LC19和LC22均为黄酮醇苷类化合物。前期研究表明[20],黄酮醇苷类在负离子模式下发生糖苷键异裂生成相应苷元子离子[A-H]-的同时,还可通过糖苷键均裂生成相应苷元自由基子离子[A-H]·-;在同样的仪器、同样的CE等参数设定下,[A-H]-和[A-H]·-的丰度比值可为推断苷元的糖基化位点提供重要参考依据。对比这5个黄酮醇苷类成分的二级质谱碎片离子信息(表2)和化学结构(图3)可以发现,3-双糖苷LC13只生成[A-H]·-m/z300,未见[A-H]-m/z301;3-糖苷LC16和LC17产生的[A-H]·-m/z300与[A-H]-m/z301的丰度比值约为1.5;8-糖苷LC19和8-葡萄糖醛酸苷LC22只生成[A-H]-m/z317,未见[A-H]·-m/z316。由上可知,在本研究的试验条件下(AB SCIEX Triple TOFTM5600质谱仪,CE为40 eV),不同糖基化位点的黄醇酮苷类生成[A-H]·-与[A-H]-的比例从大到小的排列依次为:3-双糖苷>3-糖苷>8-糖苷,这与前期文献报道[20]的排列顺序(3-糖苷>4'-糖苷>7-糖苷≈8-糖苷)基本一致。上述均裂离子产生规律对复方中其他黄酮醇苷类的化合物糖基位点归属具有重要指导意义。
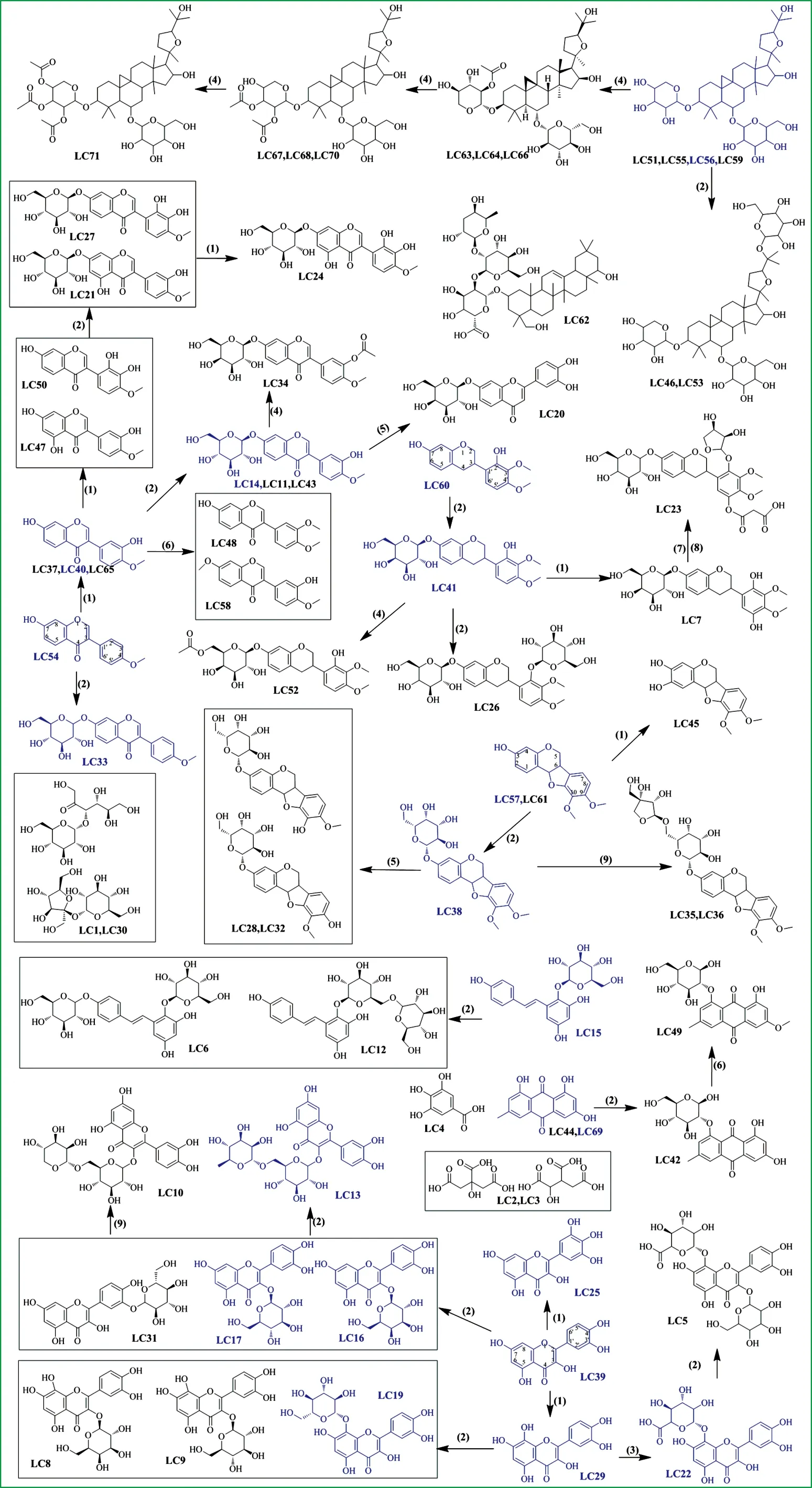
注:(1)羟基化;(2)六碳糖基化;(3)葡萄糖醛酸化;(4)乙酰化;(5)去甲基化;(6)甲基化;(7)四碳糖基化;(8)丙二酰化;(9)五碳糖基化;蓝色表示经标准品比对确认的化合物图3 芪葵颗粒中主要化学成分的可能化学结构及其可能涉及的转化反应Fig.3 The candidate structures of the identified compounds and their possible related conversion reactions
LC19的同分异构体LC8和LC9产生的子离子与LC19完全不同,它们仅产生较弱的[A-H]-m/z317,但产生显著的[A-H]·-m/z316,由此可初步推断其糖苷键可能位于3位;对比另一组同分异构体金丝桃苷(LC16)和异槲皮苷(LC17)的3位糖基分别为3-O-半乳糖苷和3-O-吡喃葡萄糖苷,类似地,暂时将LC8和LC9分别初步鉴定为棉皮素-3-O-半乳糖苷和棉皮素-3-O-吡喃葡萄糖苷。LC5可先后特征性地丢失1个葡萄糖醛酸残基(176)和1个葡萄糖残基(162),并发生苷元均裂,先后生成特征子离子m/z479、316;和LC13只生成[A-H]·-m/z300类似,只有m/z316而未见m/z317的现象提示苷元棉皮素的葡萄糖基化可能发生在3位;同时,对比LC22可知,葡萄糖醛酸化位点应为8位,据此将LC5合理鉴定为棉皮苷-3-O-葡萄糖苷。LC10与芦丁(LC13)一样,可产生显著的槲皮素苷元[A-H]·-m/z300,以及271、255等特征碎片离子,且分子式比芦丁少CH2,据此初步推测LC10也是槲皮素的3位双糖苷,将其初步鉴定为槲皮素-3-O-葡萄糖苷-戊糖苷。
LC18的分子式为C28H32O15,与毛蕊异黄酮双葡萄糖苷的分子式相符合,但其产生的子离子中未见毛蕊异黄酮的特征离子,它可以通过特征性丢失m/z194,生成可能是其苷元离子的m/z413,并可产生主要碎片离子m/z193,暂时将其归属为黄酮类,其药材来源可能为黄芪或黄蜀葵花,化学结构有待进一步深入研究。
3.2.2 皂苷类 复方中筛选出的化合物里共有14个被归属为皂苷类,均来源于黄芪药材。其中LC56可通过对照品比对准确鉴定为黄芪甲苷,LC51、LC55和LC59因与LC56具有类似的准分子离子而被简单归属为黄芪甲苷同分异构体。LC63、LC64和LC66的分子式为C43H70O15,比LC56多1个乙酰基(C2H2O),参考前期研究[7-8],将其初步归属为黄体皂苷Ⅱ同分异构体。类似地,LC67、LC68和LC70比LC56多2个乙酰基,将其初步鉴定为黄芪皂苷Ⅰ异构体;LC71比LC56多3个乙酰基,将其初步鉴定为乙酰黄芪皂苷Ⅰ;LC46和LC53比LC56多1分子葡萄糖和1分子木糖,将其初步鉴定为黄芪皂苷Ⅴ/Ⅵ/Ⅶ同分异构体;LC62被初步鉴定为大豆皂苷。
3.2.3 蒽醌类 复方中筛选出的化合物里共有4个被归属为蒽醌类,均来源于何首乌药材。其中LC69可通过标准品比对分别准确鉴定为大黄素;与文献报道[16]一致,大黄素的[M-H]-m/z269可通过先后丢失CO、CO2、CH3等生成m/z241、225、210、197、185、182等系列特征子离子。依据一级、二级质谱信息的相似性,LC44被初步鉴定为大黄素同分异构体。类似地,LC42可通过特征性地丢失1个葡萄糖苷残基生成大黄素苷元离子m/z269,苷元进一步裂解可产生m/z241、225、210等大黄素系列特征子离子,据此初步鉴定其为大黄素-8-O-β-D-葡萄糖苷。与文献报道[14,16-17]不同,复方水提液样品中未检测到大黄素甲醚,但依据特征性地162丢失,苷元离子m/z283以及m/z240等大黄素甲醚主要特征子离子(图2b和2d)的出现,LC49被合理归属为大黄素甲醚-8-O-β-D-葡萄糖苷,与文献报道相一致[18]。
3.2.4 二苯乙烯苷类 二苯乙烯苷类化合物为何首乌药材的主要化学成分之一。本研究从复方中筛选出3个二苯乙烯苷类LC6、LC12和LC15。在TOFMS负离子模式下,LC15可以产生明显而丰富的准分子离子如m/z405[M-H]-、811[2M-H]-、451[M+HCOOH-H]-、468[M+HNO3-H]-等;子离子模式下,m/z405[M-H]-可通过特征性地丢失1个葡萄糖苷残基(162)产生基峰四羟基二苯乙烯苷元离子m/z243,以及m/z225、215、173、137、131、109等苷元重排后产生的特征碎片离子,上述裂解途径及特征子离子与文献[16]中反式二苯乙烯苷的相关描述相一致;通过对照品对比,将其准确鉴定为二苯乙烯苷。LC6和LC12均可产生明显而丰富的各种准分子离子(其中LC6以[M+HCOOH-H]-m/z613丰度最高,而LC12则主要产生[M-H]-m/z567,这或许与其化学结构不同密切相关,有待深入研究考证),表明其分子式为C26H32O14,比LC15(C20H22O9)多C6H10O5,且均可产生四羟基二苯乙烯苷元特征子离子m/z243,据此初步推测其为二苯乙烯苷的葡萄糖苷;LC6的[M-H]-可产生子离子m/z405,表明LC15结构母核上发生糖基化取代的可能为酚羟基,据此初步将其归属为二苯乙烯苷-O-葡萄糖苷;而LC12的[M+HCOOH-H]-和[M-H]-的子离子中均未见m/z405的出现,表明其LC15结构母核上发生糖基化取代的可能为葡萄糖残基;据此,将LC12初步鉴定为四羟基二苯乙烯-2-O-双葡萄糖苷,其结构如图3所示。
3.2.5 其他类 LC1和LC30为一组同分异构体,均可产生丰富的准分子离子m/z341[M-H]-、387[M+HCOOH-H]-、404[M+HNO3-H]-和377[M+HCl-H]-等,但其保留时间分别为1.16 min和15.66 min,表明其极性相差较大,结构也应完全不同。参考文献报道[13]和AB Sciex数据库,暂时将其鉴定为蔗糖或松二糖,具体结构有待进一步确证。由于多糖为黄芪、黄蜀葵花和何首乌药材里面的共有主要成分,且芪葵颗粒为3味药材组成的水提制剂,据此推测芪葵颗粒中检测到的上述2种二糖来源于复方药材,而非制剂辅料。
LC2和LC3也是一组同分异构体,在TOFMS全扫描图谱上,准分子离子m/z383[2M-H]-、193[M+H]+、215[M+Na]+、210[M+NH4]+的出现,证明离子m/z191为此组同分异构体的[M-H]-,而非源内裂解碎片离子,其对应的分子式为C6H8O7,根据AB Sciex数据库匹配结果暂时将其鉴定为异柠檬酸或柠檬酸。
LC4的保留时间为1.71 min,m/z339[2M-H]-碎片的存在证明离子m/z169为其[M-H]-,而非源内裂解离子;由精确分子质量可推知其分子式应为C7H6O5,与何首乌药材中的主要成分没食子酸[8]相符;与文献报道一致[7-8],其特征性子离子m/z125、81等的出现进一步验证了该推断。
4 讨论
本研究通过全面考察库中化合物的XIC图,不仅可以发现已有报道的化合物,还能全面筛选其同分异构体和与之结构母核类似(具有相同子离子)的新化合物。化合物的锁定依据为不少于2个准分子离子的存在,且通常情况下为一个离子模式下不少于2个;与前期文献报道[11,15-16]不同,设定这样的锁定依据,可以避免将子离子对应的结构碎片鉴定为化合物的错误风险。同时,为了弥补基于已知化合物库筛选的局限性,全面系统表征芪葵颗粒复方的化学成分,本研究对复方主要化学成分(离子强度大于9 000 cps)进行了非靶向筛选,其结果既在一定程度上验证了靶向筛选的结果,也发现了一些新的未知化合物。
根据筛选鉴定出的71个化合物,作者尝试对芪葵颗粒中主要化学成分涉及到的转化反应进行总结归纳。在此过程中,假设转化反应的起点为结构最简单的化合物,即将芒柄花素、紫檀烷和异黄烷3个苷元作为黄芪黄酮类化合物的转化起点,将槲皮素、黄芪甲苷、大黄素和二苯乙烯苷分别作为黄葵黄酮类、黄芪皂苷类、何首乌蒽醌类和何首乌苯乙烯苷类化合物的转化源头。需要指出的是,上述转化起点及转化反应方向在生成复方化学成分的过程中也有可能是可逆的。如图3所示,在上述假设下,鉴定结果表明,芪葵颗粒主要化学成分的形成可能涉及羟基化、糖基化、葡萄糖醛酸化、乙酰化、甲基化、丙二酰化等多种转化反应及其逆反应。
如表2所示,对71个已鉴定化合物的统计结果显示,各种准分子离子类型出现的频率从高到低依次为:[M-H]-(58次)>[M+HNO3-H]-(54次)=[M+H]+(54次)>[M+Na]+(46次)>[M+HCOOH-H]-(44次)>[M+HCl-H]-(38次)>[M+NH4]+(28次)。此外,某些化合物也有[2M-H]-和[2M+HCOOH-H]-等二聚体准分子离子的存在。多种准分子离子的高频率同时出现为明确化合物的存在,排除源内裂解碎片离子的干扰提供了极大的便利。不过,不同类型准分子离子的产生及其丰度高低可能会受到流动相组成、化合物类型和具体的化学结构等多种因素的影响。如本研究及前期研究[7-8,10]显示:采用LC-MS系统会使多数化合物产生明显的[M+HNO3-H]-;LC流动相中甲酸和甲酸铵的加入会增加[M+HCOOH-H]-和[M+NH4]+的丰度和出现频率。因此,在中药成分定性分析中,适当优化流动相的添加成分,不仅能够改善化合物的峰形,还可以丰富准分子离子的类型,提高化合物筛选的效率和准确度。化合物的类型或结构与主要准分子离子的类型也显现出一定的相关性,本研究结果初步显示,皂苷类化合物多以[M+HCOOH-H]-为主,黄酮苷元多以[M-H]-为主,黄芪黄酮苷多以[M+HCOOH-H]-和[M+HNO3-H]-为主,而黄葵黄酮苷则以[M-H]-和[M+HNO3-H]-为主。互为同分异构体的化合物主要加合离子类型也有可能完全不同,如前面鉴定部分提到的LC6和LC12。上述加合离子的存在类型和丰度与化学结构的内在联系及其规律有待后续系统严谨的深入研究。
已鉴定化合物的峰强度信息显示,丰度最高的前15个化合物(峰强度大于100 000 cps)依次为LC15、LC2、LC3、LC40、LC54、LC1、LC69、LC22、LC17、LC16、LC14、LC33、LC4、LC31和LC38。这些成分包含何首乌中的二苯乙烯苷、没食子酸和大黄素,黄芪中的3个异黄酮苷和2个苷元,黄蜀葵花中的4个黄酮苷,以及可能存在于3种药材中的糖类和柠檬酸,这也在一定程度上提示3味药材对复方发挥药效均具有重要的作用。
综上,本研究基于UPLC-QTOF-MS/MS分析技术平台,采用靶向和非靶向相结合的筛选鉴定方法,从芪葵颗粒中筛选出89个主要化学成分,并对其中71个进行了准确鉴定或合理归属。该研究明确了芪葵颗粒中化学物质的存在形式,为科学阐明芪葵颗粒的药效物质和优化其制剂质控标准提供了重要参考依据;同时,本研究也表明基于UPLC-QTOF-MS/MS分析的靶向和非靶向相结合的筛选鉴定策略在中药等复杂化学成分的系统定性表征方面具有巨大的应用潜力。
猜你喜欢
陕西科技大学学报(2022年6期)2022-12-01
食品工业科技(2021年17期)2021-09-14
世界科学技术-中医药现代化(2021年12期)2021-04-19
食品工业科技(2020年21期)2020-11-19
天然产物研究与开发(2018年5期)2018-06-13
中山大学学报(自然科学版)(中英文)(2017年6期)2017-12-22
山西青年(2017年16期)2017-09-03
中成药(2017年6期)2017-06-13
中山大学学报(自然科学版)(中英文)(2014年4期)2014-03-27
中医研究(2014年4期)2014-03-11
