
Berberine Activates Autophagy to Induce Apoptosis of Colorectal Cancer Cells in A Caspase-Dependent Manner
2023-02-09 12:56:24-,-,-,-,-,G-,-,-,
南京中医药大学学报 2023年1期
-, -, -, -, -, G -, -, -,
(1. School of Integrated Chinese and Western Medicine, Nanjing University of Chinese Medicine, Nanjing 210023, China; 2. Collaborative Innovation Center of Jiangsu Province of Cancer Prevention and Treatment of Chinese Medicine, Nanjing University of Chinese Medicine, Nanjing 210023, China; 3. Zhangjiagang TCM Hospital Affiliated to Nanjing University of Chinese Medicine, Zhangjiagang 215000, China; 4. School of Medicine and Holistic Integrative Medicine, Nanjing University of Chinese Medicine, Nanjing 210023, China)
ABSTRACT: OBJECTIVE To investigate the potential mechanism of berberine in autophagy-mediated apoptosis of colorectal cancer. METHODS HCT116 cells and a mouse model of xenograft tumor were used to detect the tumor suppressor effect of berberine and its potential mechanism. CCK8 assay was used to investigate the cell viability. Cell cloning assay was used to detect the effect of berberine on cell growth, and the influence of berberine on cell apoptosis was evaluated by using flow cytometry assay and TUNEL staining. The transmission electron microscope and mCherry-GFP-LC3B adenovirus translocation assays were used to observe the change of autophagy in cells. HE, immunohistochemical and TUNEL staining of mouse tissue were used to estimate the effect of berberine on colorectal cancer in vivo. RESULTS Berberine inhibited the viability of tumor cells, and increased the apoptosis rate and LC3B level in HCT116 cells. 3-MA, CQ or BafA1 promoted the growth of HCT116 cells and cancelled the effect of berberine on apoptosis. Rapamycin increased the effect of berberine on up-regulating LC3B level in HCT116 cells, while Z-VAD-FMK or ATG5 siRNA abolished the effect of berberine on inducing cancer cells apoptosis. In vivo, berberine reduced tumor volume and tumor weight, and caused apoptosis in tumor tissues. In mouse tumor tissues, p-mTOR expression was significantly inhibited by berberine, while ATG5, ATG7, cleaved-caspase3 and cleaved-caspase8 were significantly up-regulated. CONCLUSION Berberine induces autophagy and promotes caspase-dependent apoptosis of HCT116 cells, thereby inhibiting cell proliferation of colorectal cancer.
KEYWORDS: berberine; colorectal cancer; autophagy; apoptosis; caspase; mouse xenograft tumor model
1 Introduction
Colorectal cancer is a common malignant tumor, ranking 2nd among female malignancies and 3rd among male malignancies, accounting for about 10% of newly diagnosed cancers worldwide each year[1]. The traditional treatment of colorectal cancer is a combination of surgery, chemotherapy, radiotherapy, and targeted therapy[2]. However, the development of therapeutic drugs for colorectal cancer is still a current research focus, and the development of new drugs with high efficiency, obvious effects, and less adverse reactions is still the main research direction at present[3].
The application of traditional Chinese herbal medicine in China has a history of thousands of years, a variety of effective ingredients are extracted from traditional Chinese herbs to treat tumors[4]. The extracts of traditional herbal medicine not only have direct therapeutic effects, but also improve other medicinal effects or reduce the incidence of adverse reactions[5-6]. Berberine (C20H19NO5), is an isoquinoline alkaloid of the traditional plantCoptischinensisFranch[7]. It has long been used as an over-the-counter drug to treat intestinal infections and diarrhea[8]. Berberine has broad anti-tumor effects. Studies have shown that its anti-tumor spectrum mainly includes lung cancer, breast cancer, nasopharyngeal carcinoma, gastric cancer, hepatoma, and colorectal cancer[9-10]. In recent years, the research on berberine has shifted frominvitrotoinvivo[11].This phenomenon indicates that the reproducibility in cell models and animal models and the transformation potential of anti-tumor effects of berberine have attracted attention.
Many studies have found that the mechanism of berberine's anti-tumor effect involves proliferation, apoptosis, migration and metastasis of tumor cells, and the mechanism is different for different types of tumors[12]. Many studies have confirmed that berberine can effectively inhibit colorectal cancerinvivoandinvitroexperiments[13], while having a minor damage to normal intestinal mucosal cells[14]. However, researches on berberine against colorectal cancer mainly focus on inhibiting the formation of tumor cells, inducing apoptosis, inhibiting proliferation and metastasis[15], and the possible mechanism of berberine inhibiting colorectal cancer still needs further exploration.
In fact, a study has found that when berberine exerts cytotoxic effects to induce tumor cell death, it not only participates in apoptosis but also accelerates autophagy[16]. In the study of gastric cancer cells, a series of characteristic morphological changes of autophagy appeared after the cells were treated with berberine, accompanied by the changes of Beclin-1 and Bcl-2, which confirmed that berberine is involved in the process of autophagy[17]. Berberine can also increase the expression of Beclin-1 in hepatoma cells, inhibit mTOR, and promote the process of autophagy and apoptosis[18]. However, due to the complexity and intersectionality of the regulatory network of apoptosis and autophagy, the specific mechanism of berberine in the process of autophagy remains to be further studied. And in colorectal cancer cells, whether berberine exerts an anti-tumor effect by regulating autophagy is also worth exploring. Therefore, in the current study, we focused on exploring whether berberine can induce apoptosis of colorectal cancer cells by regulating autophagyinvivoandinvitro. This study confirmed that berberine is an effective traditional herbal extract that inhibits the growth of colorectal cancer cells, and berberine induces apoptosis of colorectal cancer cells in a caspase-dependent manner by activating autophagy.
2 Materials and methods
2.1 Reagents
Berberine (Figure 1A), 3-Methyladenine (3-MA), chloroquine diphosphate (CQ), Bafilomycin A1 (BafA1), Z-VAD-FMK and Rapamycin (FRAP/mTOR inhibitor) were purchased from MCE. AdPlus-mCherry-GFP-LC3B was purchased from Beyotime. Control siRNA, ATG5 siRNA and transfection reagents were purchased from Santa Cruz. One-step TUNEL cell apoptosis detection kit (green fluorescence), Annexin V-FITC cell apoptosis detection kit, DAB horseradish peroxidase color kit, horseradish peroxidase labeled Streptavidin, hematoxylin eosin (HE) staining kit were purchased from Solarbio. Recombinant Anti-mTOR (phospho S2448) antibody (ab109268), recombinant Anti-Cleaved caspase-3 antibody (ab32042), ATG5 and ATG7 antibodies were purchased from Abcam. Cleaved-caspase-8 Polyclonal Antibody was purchased from ImmunoWay Biotechnology Company YC0011. Goat anti-rabbit IgG H&L (HRP) (ab205718) was from Abcam.
2.2 Cell Culture
After routine resuscitation, HCT116 cells were cultured in the 1640 culture medium supplemented with 10% fetal bovine serum and 0.5% antibiotics in a 37 ℃, 5%CO2constant temperature incubator. The HCT116 cells were treated with berberine before the detection. In different experiments, cells were treated with different concentration of berberine for different time. When HCT116 cells were incubated with 3-MA (10 mmol·L-1), CQ (20 μmol·L-1), or BafA1 (10 nmol·L-1) alone or with berberine, these reagents were administered 1 h in advance. In some tests, HCT116 cells were incubated with Z-VAD-FMK (50 μmol·L-1) and berberine for 48 h. For ATG5 silencing, according to the instructions, HCT116 cells were administrated with ATG5 siRNA and transfection reagents in the 1640 culture medium without serum and antibiotics for 6 hours, and replaced the culture medium with the 1640 culture medium supplemented with 10% fetal bovine serum and 0.5% antibiotics. After 18 hours, these transfected cells were subjected to other treatments.
2.3 CCK-8 cell viability detection
HCT116 cells were seeded in a 96-well plate, and the original medium was discarded after the cells were adhered. The corresponding concentration of berberine was prepared with the 1640 culture medium supplemented with 10% fetal bovine serum and 0.5% antibiotics. After incubating for a certain period, 10 μL of CCK-8 reagent was added to each well, and incubation for 2 hours in the incubator. Then microplate reader was used to measure the absorbance (OD) at a wavelength of 450 nm. The survival rate of untreated HCT116 cells is defined as 100%, and the survival rate is calculated according to the formula: cell survival rate=(OD value of test group/OD value of control group)×100%.
2.4 Cell cloning
HCT116 cells in the logarithmic growth phase were added to a 6-well plate and cultured in the 1640 culture medium supplemented with 30% fetal bovine serum and 0.5% antibiotics. Medium with different concentrations of berberine was added to the 6-well plate. After 48 h, all mediums were replaced with the 1640 culture medium supplemented with 30% fetal bovine serum and 0.5% antibiotics, and replaced again after three days. The culture was terminated 3 days after the second medium change. The cells were washed twice with PBS and fixed with paraformaldehyde for 10 min, then covered with an appropriate amount of Giemsa staining solution for 10 min. The excess staining solution was slowly washed off and pictures were taken. The clone formation rate is calculated according to the formula: clone formation rate=the number of inoculated cells/colony formation×100%. The relative number of clones was counted in the untreated group.
2.5 Transmission electron microscopy
The HCT116 cells were collected after being treated with berberine (20 μmol·L-1) for 6 hours, and the untreated cells were served as the control group. After fixation with glutaraldehyde, the cells are centrifuged and refixed with osmium acid. After the cells were suspended, the suspension was sucked and dropped onto a copper mesh with a membrane, the staining solution was added, and then dried. The transmission electron microscope was used to observe and take pictures.
2.6 mCherry-GFP-LC3B adenovirus translocation
According to the instructions, 20 DOI mCherry-GFP-LC3B adenovirus was added to a culture dish containing HCT116 cells, after being incubated for 12 h, the medium supplemented with 10% fetal bovine serum and 0.5% antibiotics was added for another 12 h. The cells were then collected and added to a six-well plate containing cell slides. According to the experimental requirements, the cells were added with the corresponding reagent or berberine, after a certain period of incubation, the cells are washed with PBS and fixed with 4% paraformaldehyde. After dropping the anti-fluorescence quenching mounting solution on the slide, the cell climbing slide was photographed under a fluorescence microscope.
2.7 Flow cytometry
HCT116 cells were fixed with paraformaldehyde after different treatments and the cells were collected. The cells were resuspended in PBS, counted, and centrifuged at 1000 g for 5 min. And the supernatant was discarded. Then 195 μL Annexin V-FITC binding solution, 5 μL Annexin V-FITC and 10 μL propidium iodide staining solution were sequentially added to the cells and gently mixed, which were incubated at room temperature and protected from light for 10 min and placed in an ice bath. Flow cytometry was used to detect the percentage of Annexin V-FITC and PI in 10,000 HCT116 cells.
2.8 TUNEL staining of HCT116 cells
HCT116 cells were seeded in 6-well plates with cell slides. After the cells were incubated with different concentrations of berberine for 48 h, they were fixed with 4% paraformaldehyde. The fixed cells were washed once with PBS and incubated with TUNEL staining working solution for 1 h at room temperature. After washing the cells with PBS again, DAPI staining solution was added and incubated for 5 min at room temperature. After washing the cells three times with PBS, the cell slide was taken out and placed on a glass slide dripped with anti-fluorescence quenching mounting fluid. A fluorescence microscope is used to take pictures of blue cell nuclei and green TUNEL positive expression spots.
2.9 Xenograft mouse model
BALB/c nude mice were anesthetized and injected 1×106HCT116 cells subcutaneously into the right flank. The tumor length and width were weighed and measured every day, and the tumor volume was calculated according to the formula: length×width2/2. The second day after injection, the mice in the administration group were injected with 5 mg·kg-1oxaliplatin and the 10 mg·kg-1berberine solution every day and measure the tumor volume for 8 times (0, 3, 6, 9, 12, 15, 18 and 21 days after the first administration). Meanwhile, the control mice were given normal saline. After that, all mice were sacrificed and tumors were taken out and weighed. The heart, liver, lungs, and kidneys of the mice were taken out and fixed in 4% paraformaldehyde.
2.10 HE, immunohistochemical and TUNEL staining of mouse tissue
The heart, liver, lung, kidney, and tumor tissues fixed in 4% paraformaldehyde were cut into pieces and placed in a disposable plastic embedding frame, then being dehydrated with ethanol of different concentration gradients. The dehydrated tissue was placed in xylene for 1 h, then it was soaked in melted paraffin for 1 h, replaced with paraffin and soaked again for 1 h, and then embedded into a wax block. After conventional paraffin sectioning, the wax block with tissue was baked in a 60 ℃ incubator for 30 min, soaked in xylene to dewax, and washed with gradient alcohol to remove xylene until being rehydrated with distilled water.
HE staining: Tissue sections were stained with hematoxylin for 3 min, washed with tap water for 2 min; differentiated with 1% hydrochloric acid alcohol for 20 s, washed with tap water for 5 min, and then stained with eosin for 90 s. After the gradient dehydration, the tissue sections were mounted with xylene-neutral gum mounting solution, then observed and photographed under an ordinary optical microscope.
Immunohistochemical staining: Mouse tumor tissue sections were placed in a 3% H2O2incubator, incubated at 37 ℃ for 10 min, washed with PBS for 3 times, and then antigen retrieval was performed. The tissue sections were then placed in 95 ℃ citrate buffer, incubated for 20 min, and left at room temperature for at least 20 min to cool. The tissue sections were placed horizontally in a humidifier, and 5% BSA was dripped to cover the tissue, and incubated for 20 min at room temperature. After the blocking solution was shaken off, 150 μL diluted (1∶50) p-mTOR, ATG5, ATG7, cleaved-caspase3 and cleaved-caspase8 antibodies were added to the slices to cover the tissue at 4 ℃ overnight. After washing 3 times with PBS, 150 μL of the secondary antibody was added dropwise to cover the tissue at room temperature for 1 h. After washing 3 times with PBS, 150 μL of DAB color developing liquid was added dropwise to cover the tissue, and washed with PBS after full-color development. Hematoxylin staining, dehydration, transparency, and mounting were the same as HE staining. An ordinary optical microscope was used for observation and photographing. Image pro plus was used to calculate the average optical density of the area where each antibody is positively expressed.
TUNEL staining: Mouse tumor tissue sections were dripped with TUNEL detection solution and incubated for 60 min at 37 ℃ in the dark, washed 3 times with PBS, and incubated with DAPI staining solution for 5 min at room temperature. After washing the cells 3 times with PBS, the anti-fluorescence quenching mounting solution was dropped onto the glass slide. A fluorescence microscope was used to take pictures of blue nuclei and green TUNEL positive expression spots in tumor tissues. TUNEL positive expression rate was calculated as the area of the positive expression area/the area of the entire photo.
2.11 Statistical analysis
The data were expressed as mean±SD, and the results of the data were analyzed by GraphPad Prism 6.0 software. One-way ANOVA was used for comparison between multiple groups, andP<0.05 was considered statistically significant.
3 Results
3.1 Berberine induced autophagy and inhibited cell viability in HCT116 cells
First, the cells were treated with different concentrations of berberine (10, 20, 40 μmol·L-1) for 12 hours. All three doses of berberine significantly inhibited the growth of HCT116 cells, which was confirmed by both microscope and CCK-8 cell viability experiments (Figure 1Band 1D). The appropriate concentration of berberine (20 μmol·L-1) was selected to administrate the cells for different times (0-12 hours). The results showed that after treating with 20 μmol·L-1berberine for 3 hours, the growth of HCT116 cells was significantly inhibited (Figure 1C and 1E). The IC50of berberine to inhibit the proliferation of HCT116 cells (24 h, 48 h and 72 h) were 27.01, 13.93 and 7.336 μmol·L-1(Figure 1 G-I). Meanwhile, berberine also significantly inhibited SW620 cells’ viability (Figure 1J). After that, a transmission electron microscope was used to observe the formation of autophagosomes in HCT116 cells treated with 20 μmol·L-1berberine for 6 hours. The results showed that berberine increased the number of autophagosomes in the cells (Figure 1F). To confirm this result, AAV-mCherry-GFP-LC3B transfected HCT116 cells were used. Under the same treatment, that is, after treating HCT116 cells with 20 μmol·L-1berberine for 6 hours, the number of either GPF-labeled or mCherry-labeled LC3B positive cells increased significantly (Figure 1G-H). And western bolt assay results demonstrated that berberine increased the protein level of LC3A/B (Figure 1 M, N).

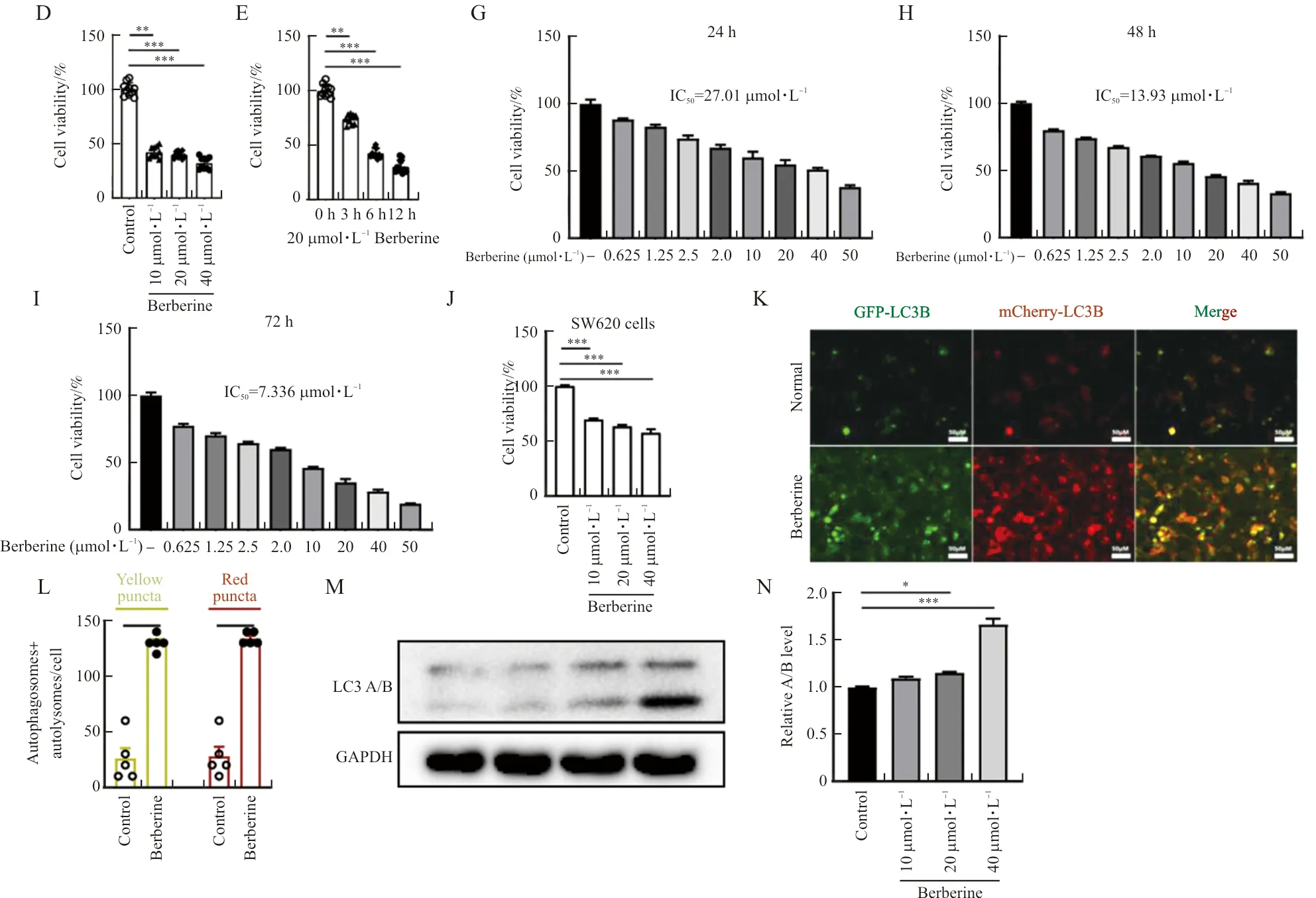
Note: (A) The chemical structure of berberine. (B, C) The cell morphology was observed under the light microscope. (D-E) The cell viability of HCT116 cells treated with berberine. (F) Autophagic vesicles of berberine-treated HCT116 cells. The red arrows indicated possible autophagosomes. (G-H) The cell viability of HCT116 cells treated with berberine. (J) The cell viability of SW620 cells treated with berberine. (K, L) LC3B positive cells and the number of GPF-LC3B or mCherry-LC3B positive cells in figure G. (M, N) Expression of LC3 A/B in berberine treated HCT116 cells. The scale bar in blue color was 1 μm. The scale bar in black and white color was 50 μm. n=10 in B-E, n=5 in F, K-L, n=3 in G-J, M, N. In the indicated comparison,*P<0.05,**P<0.01,***P<0.001.
3.2 Berberine inhibits cell viability by activating the autophagic flux in HCT116 cells
In order to determine whether the decrease in cell viability of berberine-treated HCT116 cells was caused by abnormal autophagy, three autophagy inhibitors, 3-MA (10 mmol·L-1), CQ (20 μmol·L-1) or BafA1 (10 nmol·L-1), were treated for 1 hour in advance, and then 20 μmol·L-1berberine was added to administrate for 6 hours. These autophagy inhibitors single application significantly promoted the proliferation of HCT116 cells and abolished the inhibitory effect of berberine on cell proliferation (Figure 2A, B). In AAV-mCherry-GFP-LC3B transfected HCT116 cells, 3-MA (10 mmol·L-1) blocked the initial stage of autophagy and cancelled the upregulation of berberine on LC3B-positive cells (Figure 2C-E). And autophagy inhibitors CQ (20 μmol·L-1) or BafA1 (10 nmol·L-1) inhibits the fusion of autophagosomes and lysosomes in the later stage, the separate application of the two inhibitors led to the accumulation of LC3B in cells, they promoted cell proliferation while also promotes the up-regulation of berberine on the positive expression of LC3B in HCT116 cells (Figure 2C-E). When the autophagy inducer rapamycin (10 nmol·L-1) was applied to HCT116 cells alone or together with 20 μmol·L-1berberine for 6 hours, the number of HCT116 cells that were either GPF-labeled or mCherry-labeled LC3B-positive increased significantly (Figure 2F-H). And compared with cells using rapamycin or berberine alone, the combination of the two has a more significant effect on the promotion of LC3B expression in HCT116 cells (Figure 2E-G).
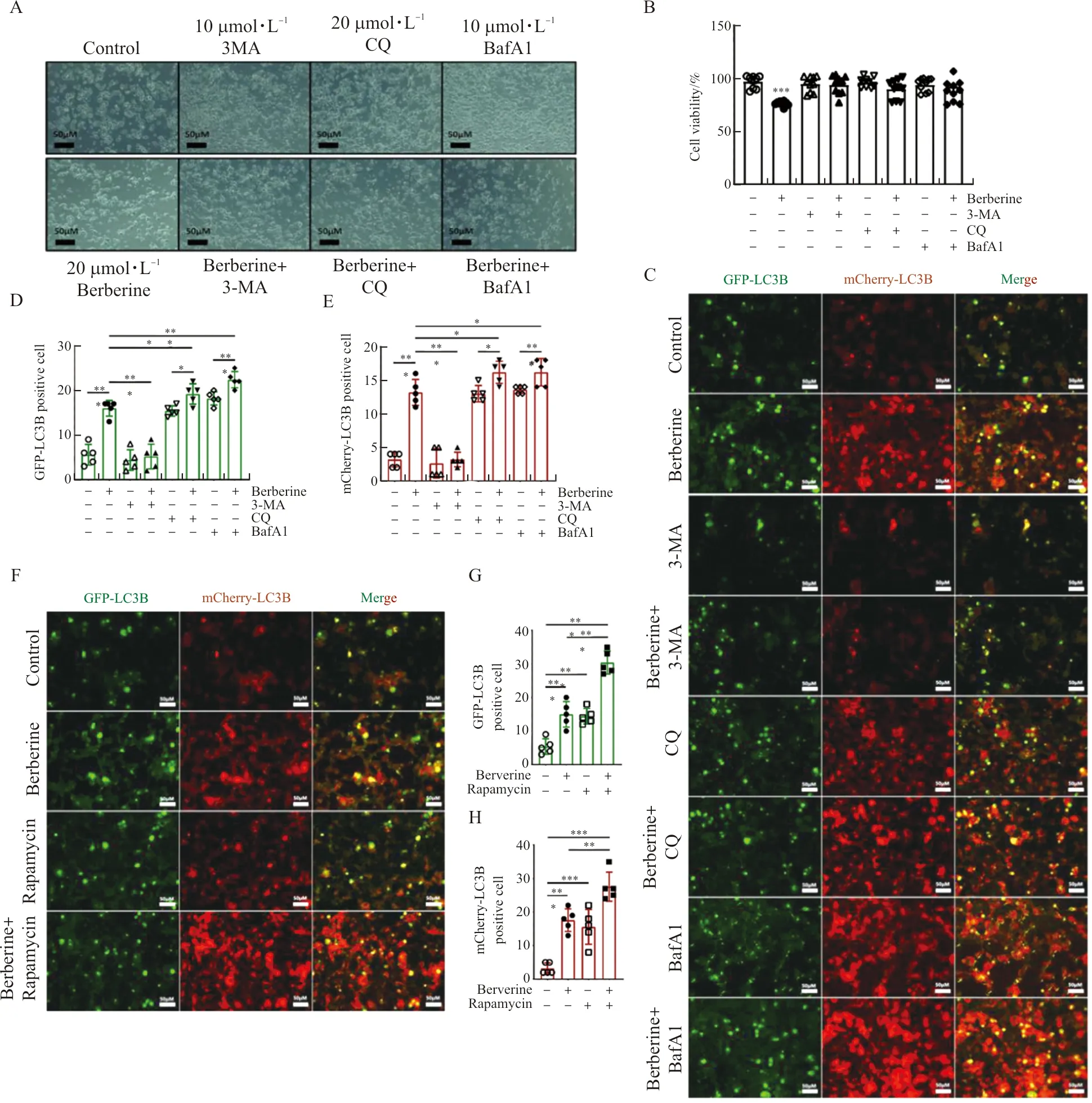
Note: (A, B) Cell morphology and viability of HCT116 cells were pre-treated with 3-MA (10 mmol·L-1), CQ (20 μmol·L-1) or BafA1 (10 nmol·L-1) for 1h and then treated with or without berberine (20 μmol·L-1) for 6 h. (C-E) The AAV-mCherry-GFP-LC3B transfected HCT116 cells were pre-treated with 3-MA (10 mmol·L-1), CQ (20 μmol·L-1) or BafA1 (10 nmol·L-1) for 1 h and then treated with or without berberine (20 μmol·L-1) for 6 h, and the number of GPF-LC3B positive cell in C and mCherry-LC3B positive cell in C were determined. (F, G) The AAV-mCherry-GFP-LC3B transfected HCT116 cells were treated with Rapamycin (10 nmol·L-1) and with or without berberine (20 μmol·L-1) for 6 h, the number of GPF-LC3B positive cells in E and the number of mCherry-LC3B positive cell in E were determined. The scale bar in black and white color was 50 μm. n=10 in A, n=5 in B-G. In the indicated comparison,*P<0.05,**P<0.01,***P<0.001.
3.3 Berberine induced caspase-dependent apoptosis in HCT116 cells
Berberine not only inhibited the viability of HCT116 cells (Figure 1A-D), but the 8-day HCT116 cell cloning experiment also showed that berberine (10, 20, 40 μmol·L-1) significantly inhibited the formation of cell clones in the first 48 hours after treatment (Figure 3A-B). The apoptosis rate of HCT116 cells after berberine (10, 20, 40 μmol·L-1) treatment for 48 hours was tested by flow cytometry. The results showed that berberine significantly promoted the apoptosis of HCT116 cells (figure 3C-D). The effect of berberine in promoting HCT116 cell apoptosis was observed again with TUNEL staining. The results showed that berberine (10, 20, 40 μmol·L-1) significantly increased the number of TUNEL-positive stained cells (Figure 3I). However, when Z-VAD-FMK (50 μmol·L-1) and berberine (20 μmol·L-1) were administrated to HCT116 cells together, the inhibitory effects of berberine on clone formation and the promotion of apoptosis were abolished (Figure 3E-H). These results indicated that berberine induced autophagy in HCT116 cells and promoted cell apoptosis, reducing cell viability and colony formation, and the role of berberine depended on the normal function of caspase.

Note: (A, B) Images of the cell cloning experiment were taken after berberine was applied to the HCT116 cell, and the colony number in A was counted. (C, D) Flow cytometry was performed after berberine was applied to HCT116 cells, and the apoptosis rate was based on the results of C. (E, F) Images of the cell cloning experiment were taken after HCT116 cells were treated with or without berberine and Z-VAD-FMK for 48 h and the colony number of E were counted. (G, H) Flow cytometry was performed after HCT116 cells were treated with or without berberine and Z-VAD-FMK for 48 h and the apoptosis rate was calculated based on the results of G. (I) Tunel staining was performed after HCT116 cells were treated with or without berberine and Z-VAD-FMK for 48 h. The scale bar in white color was 50 μm. n=5. In the indicated comparison,***P<0.001.
3.4 Berberine induced apoptosis depended on autophagy in HCT116 cells
To confirm whether berberine induced apoptosis in HCT116 cells was dependent on autophagy, CQ and berberine were co-treated to HCT116 cells to detect the formation of cell clones and the rate of apoptosis. The result of the 8-day HCT116 cell clone experiment showed that the co-application of CQ (20 μmol·L-1) and berberine (20 μmol·L-1) failed to inhibit the formation of cell clones in the first 48 hours after administration (figure 4A-B). The inhibitory effect of berberine on apoptosis was also cancelled by the co-application of CQ (Figure 4C-D). To further confirm this result, HCT116 cells were transfected with ATG5 siRNA. In HCT116 cells where ATG5 was silenced, treatment with berberine (20 μmol·L-1) failed to inhibit the formation of cell clones, nor did it induce apoptosis of HCT116 cells (Figure 4E-H-D). These results confirmed that berberine induced apoptosis in HCT116 cells depends on the autophagy function of cells.

Note: (A, B) Images of the cell cloning experiment were taken after HCT116 cells were treated with or without berberine and CQ and the colony number were counted. (C, D) Flow cytometry was performed after HCT116 cells were treated with or without berberine and CQ, and the apoptosis rate was calculated based on the results of C. (E, F) Images of the cell cloning experiment were taken in HCT116 cells with control siRNA or ATG5 siRNA transfection followed with or without berberine treatment, and the colony number was counted. (G) Flow cytometry was performed in HCT116 cells with control siRNA or ATG5 siRNA transfection followed with or without berberine and the apoptosis rate was calculated based on the results of G. n=5. In the indicated comparison,***P<0.001.
3.5 Berberine promoted autophagy dependent apoptosis in HCT116 xenograft tumor mice
To determine the antitumor effect and mechanism of berberineinvivo, BALB/c nude mice were injected with HCT116 cells subcutaneously. In the process of daily intraperitoneal injection of model mice with berberine (10 mg·kg-1), the weight and tumor volume of the mice were tested every three days. The results showed that no matter whether it was treated with berberine or the positive drug oxaliplatin (5 mg·kg-1), within 21 days, there was no significant difference in the body weight of model mice (Figure 5A). The tumor volume of model mice treated with berberine or oxaliplatin decreased significantly from the 9th day of treatment (Figure 5B). After 21 days of treatment with berberine or oxaliplatin, the weight of the tumors in the mice was significantly reduced (Figure 5C). These results indicate that berberine can inhibit the growth of colorectal cancer cellsinvivo. HE results showed that berberine or oxaliplatin treatment caused cell damage in the tumor tissue of model mice, but had no significant toxic effect on the heart, liver, lung, and kidney of model mice (Figure 5D). Further, the results of immunohistochemistry showed that berberine or oxaliplatin treatment inhibited the expression of p-mTOR in tumor tissues, and significantly promoted the expression of ATG5, ATG7, cleaved-caspase3 and cleaved-caspase8 (Figure 5E-J). These results indicated that berberine promoted autophagy and apoptosis in tumor tissues of xenograft mice. Moreover, TUNEL staining results showed that berberine or oxaliplatin treatment increased the area of TUNEL-positive area (Figure 5K-L), confirming that berberine promoted cell apoptosis in tumor tissues of xenograft mice.
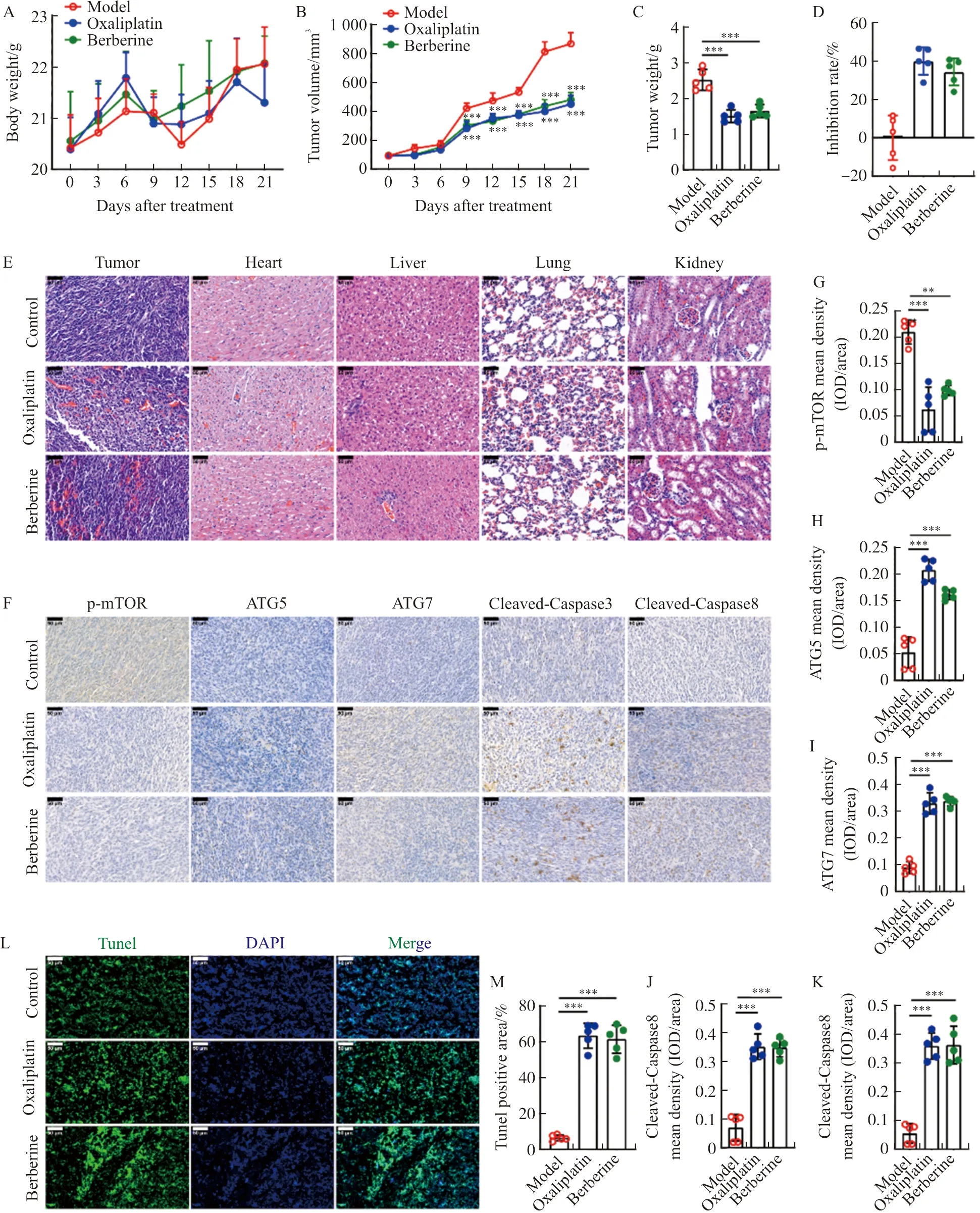
Note: (A, B) The body weight and tumor volume of HCT116 xenograft tumor mice with oxaliplatin or berberine administration. (C) The tumor weight of HCT116 xenograft tumor mice with oxaliplatin or berberine treatment. (D) The tumor growth inhibition rate of berberine. (E)HE staining was performed in tumor, heart, liver, lung, and kidney from HCT116 xenograft tumor mice with oxaliplatin or berberine treatment. (F-K) Immunohistochemical staining of p-mTOR, ATG5, ATG7, cleaved-caspase3, and cleaved-caspase8 was performed in tumors from HCT116 xenograft tumor mice with oxaliplatin or berberine treatment. And the level of proteins in tumor from mice were calculated based on the result of E. (L, M) Tunel staining was performed in tumor from HCT116 xenograft tumor mice with oxaliplatin or berberine treatment., and Tunel positive area in tumor from mice were calculated based on the result of K. The scale bar in black and white color was 50 μm. n=5. In the indicated comparison,**P<0.01,***P<0.001.
4 Discussion
The effective ingredients of traditional medicinal plants have become an important source for the development of new anti-cancer drugs[19]. Berberine has become a research hotspot due to its extensive pharmacological activity and effective inhibition of tumor cells[20]. Studies have reported that berberine has anti-colorectal cancer effects, including blocking the cell cycle in G0/G1 phase and inhibiting the proliferation of intestinal cancer cells[21].However, its mechanism remains unclear. In the current study, we provided direct evidence that berberine can inhibit HCT116 cell viability and promote autophagy. We confirmed that autophagy activation was the key link for berberine to inhibit the viability of HCT116 cells, and the activation of autophagy by berberine promoted the apoptosis of HCT116 cells in a caspase-dependent manner. In the xenograft mouse model, berberine inhibited tumor growth by inducing autophagy to promote the apoptosis of colorectal cancer cells.
Increased studies on anti-tumor drugs have shown that inhibiting tumor proliferation is mainly achieved by inducing tumor cell autophagy and apoptosis[22]. Autophagy and apoptosis are two forms of programmed cell death, the former is usually regarded as an active death process, while the latter is a passive cell death process. Some studies suggest that autophagy may be the early stage of the apoptosis process, when autophagy reaches a certain level, it will induce cell apoptosis[23]. In this study, HCT116 cells were treated with berberine. Cell viability detection and cell cloning experiments were used to monitor cell growth. Autophagosomes were observed by transmission electron microscopy, and HCT116 cells were transfected with mCherry-GFP-LC3B adenovirus to observe autophagic flux. These experiments confirmed that berberine induced autophagy and inhibited cell viability in HCT116 cells. And the results of flow cytometry and TUNEL staining of HCT116 cells showed that autophagy induced by berberine promoted cell apoptosis. More importantly, under the intervention of a variety of autophagy inhibitors, including 3-MA, CQ and BafA1, the inhibitory effect of berberine on HCT116 cell viability was cancelled. It should be mentioned that caspase is the core factor of apoptosis, and the application of its inhibitor cancelled the inhibition of berberine on the colonization of colorectal cancer cells and the induction of apoptosis. In summary, our results confirmed that berberine induced autophagy and promoted the apoptosis of HCT116 cells in a caspase-dependent manner, thereby inhibiting cell proliferation and playing an anti-colorectal cancer effect.
So far, it has been found that the whole process of autophagy is strictly regulated by more than 36 autophagy-related genes (ATGs) and corresponding proteins[24]. The mammalian target of rapamycin (mTOR) is related to cell proliferation, stress, and cancer progression, and is a key factor for autophagy[25]. Recent studies have shown that autophagy is related to cancer initiation and treatment[26-27]. Autophagy is widely involved in the development of colorectal cancer. When the expressions of autophagy-related proteins LC3B, ATG5 and Beclin1 are lacking, the prognosis of colorectal cancer patients is often poor[28]. Among colorectal cancers with high microsatellite instability, 27% of cancer patients contain at least one mutation in the ATG2B, ATG5, ATG9B, and ATG12 genes, and decreased protein expression is associated with colorectal cancer and gastric cancer[29]. Studies have found that berberine can induce apoptosis in lung cancer by regulating the expression of Bcl-2, up-regulating the mRNA levels of Beclin1 and ATG5, and promoting autophagy to exert anti-tumor effects[30]. And some studies have confirmed that berberine can activate AMPK, inhibit the downstream target mTOR and NF-κB signaling pathways, inhibit the expression of 4E binding protein (4EBP1), and regulate the important metabolic processes such as cancer energy metabolism and protein synthesis of colorectal cancer cells in the process of preventing the occurrence and development of colorectal cancer.[31]. In the current study, we not only provided evidence that berberine promoted apoptosis of colorectal cancer cells by inducing autophagyinvitro, also confirmed that mTOR in tumor tissue was significantly inhibited by berberine in the xenograft mice model. Furthermore, we found that the silence of ATG5 abolished the induction of berberine on the apoptosis of HCT116 cells and the inhibition of clone formation.
In general, non-selective autophagy is used for nutrient stress, while selective autophagy is used for cell maintenance. It is undeniable that in the context of colorectal cancer tumor growth, these roles may change[32]. The most important research on autophagy in colorectal cancer mainly focuses on molecules such as LC3, Beclin1, ATG5 and ATG7, and many studies have produced conflicting results[33]. Some studies have shown that autophagy is a regulator of multiple oncogenes and tumor suppressor genes, while other studies have shown that autophagy is involved in not only promoting the occurrence of tumors, but also the development and suppression of cancer[34]. The exact mechanism of this contradiction has not been thoroughly studied. Although in the current study, we have confirmedinvivothat berberine promoted the apoptosis of colorectal cancer cells by inducing autophagy, which is mainly related to LC3B, ATG5, mTOR and ATG7, we have not explored more autophagy genes. We believe that it is necessary to determine how the dual role of autophagy regulates the occurrence and development of colorectal cancer, and determine what signals and molecular mechanisms make autophagy promote in one case and inhibit it in another. Although some clinical drugs have been developed for autophagy, further researches are still needed to better understand the relationship between colorectal cancer and autophagy, and to develop drugs with potential benefits for treatment and prognosis.
Although there is currently a study reporting that berberine is used in clinical practice to prevent colorectal adenoma recurrence[35], the current research on berberine inhibiting colorectal cancer and other malignant tumors is mainly concentrated in the laboratory stage, with little evidence and support from clinical trials. An additional consideration is that although berberine can be used as a treatment for tumors, the application of berberine still faces many problems, such as poor water solubility, low intestinal absorption, and low oral bioavailability[36-37]. Therefore, the application of berberine in colorectal cancer and other tumors needs further research. Studies have shown that synthetic berberine derivatives have a stronger inhibitory effect on tumor cell proliferation than berberine itself[38]. It is believed that with the continuous in-depth research on berberine combined with clinical effects and responses, it is expected to become an effective anti-tumor drug.
In summary, berberine, as an isoquinoline alkaloid with a wide range of pharmacological effects, has good anti-colorectal cancer proliferation effects bothinvivoandinvitro. Berberine activates autophagy to induce apoptosis of colorectal cancer cells in a caspase-dependent manner, thereby inhibiting the proliferation of HCT116, and might be used as a new treatment candidate for colorectal cancer.
