
抑制HDAC11减轻OGD/R所致的HT22细胞损伤*
2023-02-07 08:35郑琪雪费慧芝杨俊卿
中国病理生理杂志 2023年1期
郑琪雪 , 杨 洋 , 费慧芝 , 杨俊卿 △
(1重庆医科大学,重庆 400016;2重庆医科大学附属妇女儿童医院,重庆 400021;3重庆市妇幼保健院,重庆 400021)
新生儿缺氧缺血性脑损伤(hypoxic-ischemic brain damage, HIBD)是由围产期胎儿因缺乏血流或气体交换障碍导致[1]。新生儿的大脑需要高水平的氧气供应,因此对缺氧极为敏感。严重的HIBD导致诸多神经系统后遗症,包括智力迟钝,认知障碍,脑瘫和癫痫等[2]。目前,临床上主要采用亚低温治疗、对症治疗和营养支持等方法。HIBD的病理生理机制并不完全清楚,临床治疗效果并不理想,因此需深入探究其发展机制,寻找HIBD干预新靶点。
组蛋白脱乙酰酶(histone deacetylases, HDACs)包含4类,家族成员众多,发挥的功能作用并不相同。抑制Ⅰ类HDACs改善APP/PS1转基因小鼠的β-淀粉样蛋白沉积[3];抑制Ⅱ类HDACs调节组蛋白H2A及α-微管蛋白乙酰化水平改善脑缺血大鼠神经元损伤[4-5];与Ⅰ类和Ⅱ类作用不同,Ⅲ类HDACs可能有神经保护作用,抑制Sirt1(Ⅲ类)会加重小鼠脑缺血损伤,其机制与P5及P65乙酰化水平增加有关[6-7];HDAC11属于第Ⅳ类中唯一的成员,在脑内表达丰富,影响小鼠脑内神经细胞的发育,HDAC11是一种涉及多种细胞途径的酶,但关于所涉及的分子机制仍不清晰[8],且HDAC11在不同环境中的调节作用呈现差异性。虽有研究表明[9],抑制HDAC11促进抗炎因子白细胞介素10(interleukin-10,IL-10)的表达,但另有研究显示,敲除HDAC11基因会增加嗜中性粒细胞促炎因子肿瘤坏死因子α(tumor necrosis factor alpha, TNF-α)及IL-6的表达[10]。HDAC11在中枢神经系统疾病的作用研究尚少,HDAC11在HIBD中是否发挥作用,是保护作用还是损伤性作用,其作用机制如何,这些均不清楚,值得研究。
氧化应激是氧化剂与抗氧化剂平衡的紊乱。由于新生儿的特性,更容易受到氧化应激损伤。新生儿大脑富含脂肪酸,容易受到自由基和脂质过氧化损伤,未成熟脑组织对自由基高度敏感且抗氧化功能不完善[11]。氧化应激是HIBD发生后神经元死亡的重要原因。HIBD发生后,细胞内Ca2+、Na+和二磷酸腺苷(adenosine diphosphate, ADP)升高导致线粒体产生有害活性氧,导致多不饱和脂肪酸氧化成丙二醛(malondialdehyde,MDA),同时活性氧介导分子信号传导,其中转录因子p53激活产生促凋亡蛋白如BAX,促进坏死和细胞凋亡[12]。超氧化物歧化酶(superoxide dismutase, SOD)及还原型谷胱甘肽(glutathione, GSH)保护细胞免受自由基的侵害,内源性抗氧化物GSH的含量的增加减轻神经损伤[13]。MDA,SOD,GSH是氧化应激的标志性物质。
小鼠HT22细胞氧糖剥夺/复糖复氧(oxygen-glucose deprivation/reoxygenation, OGD/R)能够部分模拟HIBD的病理生理过程变化[14],因此本研究旨在观察N'-十六烷基噻吩-2-碳酰肼(N'-hexadecylthiophene-2-carbohydrazide, SIS17)特异性抑制 HDAC11对OGD/R致小鼠HT22细胞损伤的作用,并从酪氨酸激酶(Mer tyrosine kinase, MerTK)调控氧化应激损伤和凋亡通路初步探讨其机制。
材料和方法
1 细胞
小鼠海马区神经元HT22细胞系购自上海中乔新舟生物科技有限公司。
2 主要试剂
DMEM高糖培养液、DMEM无糖培养液及0.25%胰蛋白酶购自Gibco;青霉素-链霉素溶液(C0222)购自上海碧云天生物技术有限公司;胎牛血清购自苏州依科赛生物科技有限公司;HDAC11特异性抑制剂SIS17(HY-128918)购自MedChemExpress;LDH(A020-2),SOD(A001-3),GSH(A006-2-1)和MDA(A003-1)试剂盒均购自南京建成生物工程研究所有限公司;细胞凋亡检测试剂盒(MA0220)购自大连美仑生物技术有限公司;抗β-Tubulin抗体(10068-1-AP)购自武汉三鹰生物技术有限公司;抗HDAC11抗体(bs-2894R)及抗 caspase12抗体(bs-23014R)购自北京博奥森生物技术有限公司; 抗MerTK抗体(DF7344),抗ATF6抗体(DF6009)及抗Bax抗体(AF0120)均购自Affinity;HRP标记山羊抗兔IgG购自北京兰杰柯科技有限公司。
3 实验仪器
细胞培养箱(311,Thermo Scientific);三气培养箱(3423,Thermo Scientific);低温离心机(Icen-24R,Thermo Scientific);垂直电泳仪(041BR304570,BIORAD);成像仪(VLBL00D2, BIO-RAD)。
4 实验方法
4.1 细胞培养 完全培养液:10%胎牛血清,1%青霉素链霉素溶液,89%DMEM高糖培养液。细胞传代:HT22细胞于37 ℃培养箱中培养,密度达到85%时进行传代,弃置原完全培养液,PBS润洗2次后加入0.25%胰蛋白酶于培养箱中消化1 min,添加适量完全培养液终止消化,吹打细胞成均一悬液,1 000 ×g离心4 min,取新配置完全培养液重悬细胞,接种至新的培养瓶中。
4.2 OGD/R模型建立及实验分组 培养HT22细胞至85%密度时,除对照(control)组,其他各组弃去培养液后用PBS润洗2次,添加工作体积DMEM无糖培养液并置于三气培养箱(95% N2,1%O2,4% CO2)中进行氧糖剥夺4 h,再更换为DMEM高糖完全培养液于普通培养箱复糖复氧24 h。根据本实验对HDAC11特异性抑制剂SIS17安全范围筛查以及文献报道SIS17具有剂量依赖性[15],本研究对SIS17剂量 的 设 置 为(SIS17-L:3×10-6mol/L,SIS17-M:1×10-5mol/L,SIS17-H:3×10-5mol/L)。本次研究中细胞实验分为5组:对照(control)组,模型(OGD/R)组,OGD/R+L组,OGD/R+M组和OGD/R+H组。
4.3 MTT法检测细胞活力 HT22细胞以每孔3 000个接种于96孔板,高中低剂量SIS17处理24 h后进行OGD/R,每孔加入20 μL MTT溶液(5 g/L),37 ℃孵育4 h,低速离心,弃去培养液,每孔加入150 μL DMSO溶解甲瓒,于水平摇床避光震荡15 min充分溶解,酶标仪490 nm处测定吸光度(A)值,根据公式计算细胞活力,细胞活力(%)=[A值(实验组)-A值(空白组)]/[A值(对照组)-A值(空白组)]×100%,实验组:OGD/R,OGD/R+L,OGD/R+M,OGD/R+H;对照组:control;空白组:含完全培养液无细胞。
4.4 细胞LDH测定 收集各组细胞培养液,依次加入双蒸水,0.2 mol/L丙酮酸标准液,待测样本,基质缓冲液,辅酶I,混匀。37 ℃温浴15 min,加入2,4-二硝基苯肼,混匀,37 ℃温浴15 min,加入0.4 mol/L NaOH溶液混匀,室温放置5 min,波长450 nm,酶标仪测定吸光度值。
4.5 流式细胞术检测细胞凋亡率 将HT22细胞用不含EDTA的胰酶消化,1 000 ×g离心5 min,收集细胞,加入预冷PBS溶液洗涤离心2次,在细胞沉淀中加入结合缓冲液重悬细胞,加入5 μL Annexin VFITC和10 μL碘化丙碇(PI)染色液并混匀,室温避光孵育15 min,使用流式细胞仪检测细胞凋亡率。
4.6 TUNEL染色 用PBS润洗HT22细胞,4%多聚甲醛室温固定15 min,PBS润洗,0.3% Triton X-100室温反应30 min,PBS润洗,加入平衡缓冲液,室温反应40 min,倒掉液体加入缓冲液,37 ℃孵育1.5 h,PBS润洗后,加入DAPI溶液复染10 min,PBS润洗后,荧光显微镜下成像。200倍下选择4个具有代表性的视野,计数阳性细胞数和阴性细胞数,阳性细胞百分率%=阳性细胞总数/(阳性细胞总数+阴性细胞总数)×100%,取平均值。
4.7 SOD,GSH和MDA测定 收集各组细胞培养液,根据试剂盒说明书,参考文献方法[16],于孔板加入相应试剂,分别于450 nm和405 nm处测定SOD及GSH吸光度值。TBA法测定MDA,PBS润洗并收集细胞,破碎细胞后根据试剂盒步骤操作,于532 nm处测定MDA吸光度值。
4.8 Western blot检测蛋白表达水平 收集细胞总蛋白,加入蛋白裂解液(RIPA∶PMSF∶磷酸酶抑制剂=100∶1∶2)经破碎后低温离心(4 ℃,12 000 ×g)15 min,取上清测蛋白浓度,按1∶4的比例加入5×蛋白上样缓冲液,98℃变性15 min,取30 μg进行上样。蛋白经电泳(90V,30 min;120 V,60 min)转膜(250 mA,100 min)后经5%脱脂奶粉封闭2 h,孵育Ⅰ抗4 ℃过夜,TBST洗3次每次5min,室温孵育Ⅱ抗(1∶2 000),TBST洗3次,ECL发光液用于成像。抗体稀释倍数:兔多克隆抗体:HDAC11(1∶1 000),MerTK(1∶1 000),Bax(1∶1 000),caspase-12(1∶1 000)和 β-Tubulin(1∶1 000),HRP 标记山羊抗兔 IgG(1∶4 000),Image Lab对图像进行灰度值分析。
5 统计学处理
使用GraphPad Prism7.0软件进行统计学分析。计量数据均采用均数±标准误(mean±SEM)表示。多组间比较采用单因素方差分析。P<0.05具备统计学差异。
结 果
1 HDAC11特异性抑制剂SIS17在HT22细胞上的安全范围
MTT法筛选HDAC11特异性抑制剂SIS17的安全范围。在10-4~10-13mol/L范围内,SIS17对HT22细胞活力的影响不显著(P>0.05),见图1A。OGD/R处理HT22细胞后,细胞数量减少,呈现损伤状态;不同浓度SIS17处理OGD/R细胞后,细胞数量增加,细胞状态得到改善,见图1B。
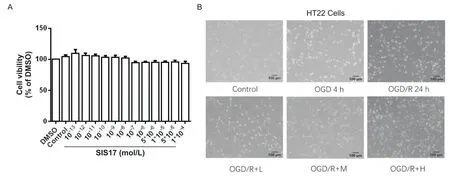
Figure 1. Effect of HDAC11 inhibitor on viability and morphological of HT22 cells treated with OGD/R. A: cell viability detected by MTT assay; B: morphological alterations of HT22 cells. Mean±SEM. n=4.图1 抑制HDAC11对小鼠HT22细胞活力及形态学的影响
2 抑制HDAC11对OGD/R处理小鼠HT22细胞的存活率及LDH影响
MTT实验结果显示,相比于control组,OGD/R组小鼠HT22细胞的活力显著降低(P<0.05);相比于OGD/R组,OGD/R+L组细胞活力有上升趋势但无显著性差异(P>0.05),OGD/R+M及OGD/R+H组细胞活力显著升高(P<0.05),见图2A。如图2B所示,相比于control组,OGD/R组HT22细胞LDH活力显著升高(P<0.01);相比于 OGD/R 组,OGD/R+L组,OGD/R+M组及OGD/R+H组HT22细胞LDH活力均显著降低(P<0.05,P<0.01)。
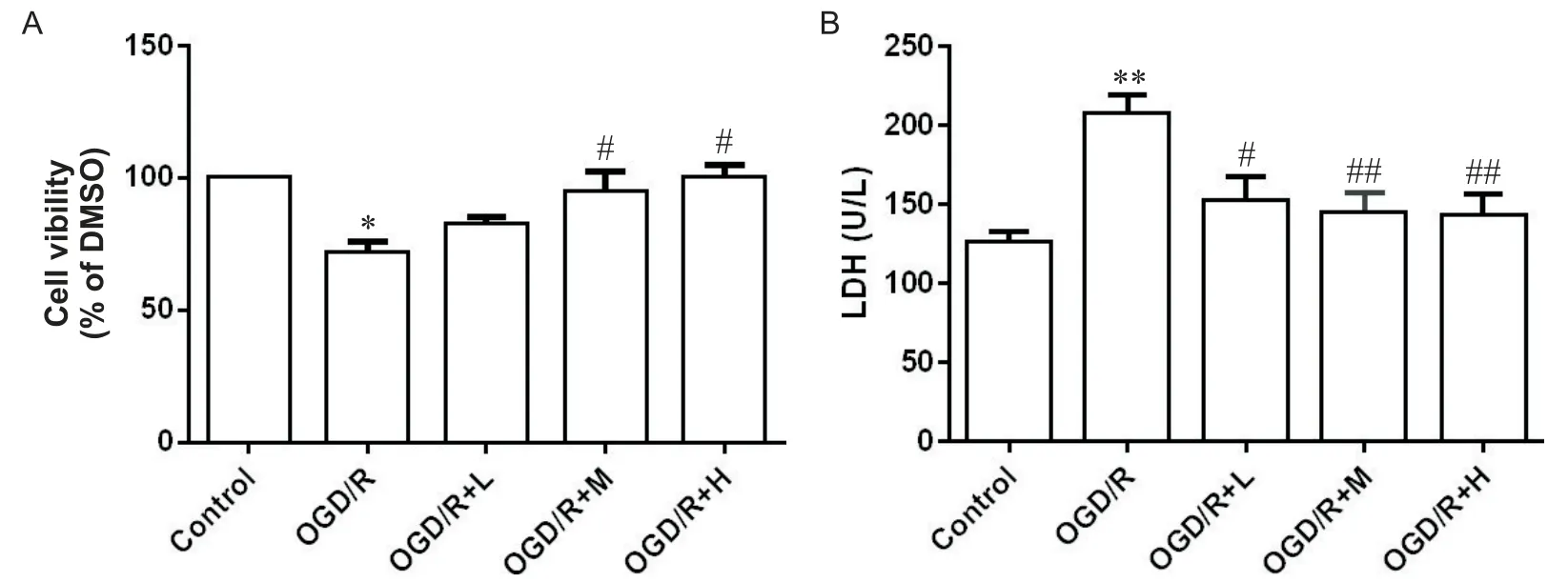
Figure 2. Effect of inhibiting HDAC11 on viability and LDH of HT22 cells treated with OGD/R. A: cell viability; B: LDH activity of HT22 cells; Mean±SEM. n=4. *P<0.05, **P<0.01 vs control group; #P<0.05, ##P<0.01 vs OGD/R group.图2 抑制HDAC11对小鼠HT22细胞活力及LDH活力的影响
3 抑制HDAC11对OGD/R处理的HT22细胞凋亡的影响
为探究HDAC11特异性抑制剂SIS17对OGD/R处理小鼠HT22细胞凋亡的影响,流式细胞术结果显示,相比于control组(凋亡率6.71%),OGD/R组细胞凋亡率升高(凋亡率37.14%),相比于OGD/R组,OGD/R+L,OGD/R+M及OGD/R+H组细胞凋亡率降低(分别为:26.69%,16.64%,15.78%)(P<0.05,P<0.01,见图3A)。如图3B,TUNEL染色结果显示,相比于control组,OGD/R组细胞凋亡率显著升高(P<0.01);相比于OGD/R组,OGD/R+H组细胞凋亡率显著降低(P<0.05)。
4 抑制HDAC11减轻OGD/R处理的HT22细胞氧化应激损伤
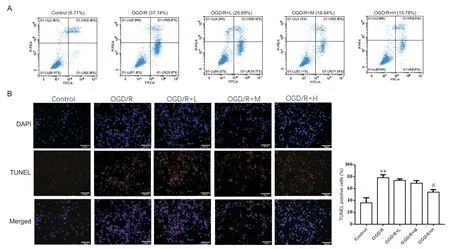
Figure 3. Inhibition of HDAC11 ameliorate apoptosis of HT22 cells damaged by OGD/R. A: the apoptosis rate of HT22 cells was detected by flow cytometry; B: TUNEL staining, blue DAPI, red TUNEL (scale bar=100 μm). Mean±SEM. n=3. **P<0.01 vs control group; #P<0.05 vs OGD/R group.图3 抑制HDAC11改善小鼠HT22细胞凋亡
为探究HDAC11特异性抑制剂SIS17对OGD/R致小鼠HT22细胞氧化应激的影响。如图4所示,相比于control组,OGD/R组超氧化物歧化酶(SOD)及谷胱甘肽(GSH)含量显著降低(P<0.01,P<0.05),丙二醛(MDA)显著升高(P<0.01),细胞氧化应激损伤明显。相比于OGD/R组,OGD/R+L组和OGD/R+M组细胞的SOD及GSH含量有上升趋势,MDA有降低趋势但无统计学(P>0.05);相比于OGD/R组,OGD/R+H组SOD及GSH含量显著升高(P<0.05),MDA含量显著降低(P<0.05)。
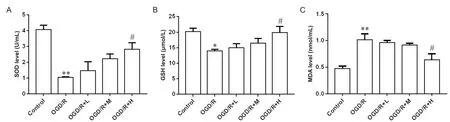
Figure 4. Inhibition ofHDAC11 ameliorate oxidative stress injury in HT22 cells damaged by OGD/R.A: SOD level; B: GSH level;C: MDA level. Mean±SEM. n=4.*P<0.05,**P<0.01 vs control group; #P<0.05 vs OGD/R group.图4 抑制HDAC11改善小鼠HT22细胞氧化应激损伤
5 抑制HDAC11对OGD/R处理的小鼠HT22细胞蛋白表达的影响
如图5所示,相比于control组,OGD/R组HDAC11,MerTK,caspase-12及Bax蛋白表达显著上调(P<0.05,P<0.01)。相较于OGD/R组,OGD/R+L组及OGD/R+M 组Bax表达显著降低(P<0.05,P<0.01),而HDAC11,MerTK和caspase-12蛋白表达水平 均 无 显 著 性 差 异(P>0.05);OGD/R+H 组HDAC11,caspase-12及 Bax显著降低(P<0.05,P<0.01),而MerTK蛋白表达显著上调(P<0.05)。
讨 论
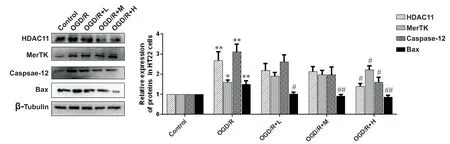
Figure 5. Effect of HDAC11 inhibitor on protein expression of HDAC11, MerTK, caspase12, BAX in OGD/R treated with HT22 cells. Mean±SEM. n=3. *P<0.05, **P<0.01 vs control group; #P<0.05, ##P<0.01 vs OGD/R group.图5 抑制HDAC11对小鼠HT22细胞蛋白表达的影响
新生儿缺氧缺血性脑损伤是(HIBD)导致死亡和终身残疾的主要原因,急需寻求新的治疗靶点。小鼠HT22细胞是一种原代小鼠海马神经元培养物中永生化的亲本HT4细胞的亚系,可以很好的体外模拟神经元功能。HDACs抑制剂多为非选择性抑制多个HDAC,不能针对性抑制特定的靶标。而SIS17是一种靶向性HDAC11抑制剂,并不影响其他HDACs的活性及表达量[14]。本研究在小鼠HT22细胞上模拟HIBD疾病模型,并初步探讨抑制HDAC11对HIBD的作用及机制。
研究表明,HIBD的发生导致神经递质释放异常,小胶质细胞受到刺激产生大量氧自由基,这些物质通过氧化还原信号破坏血脑屏障完整性,启动细胞凋亡,凋亡的细胞得不到及时清除会释放细胞因子引起更强的级联反应,从而导致神经元的进一步损伤[17-18]。新生儿因自身特性,更易受到凋亡及氧化应激损伤,MerTK通路可抑制先天免疫反应并促进凋亡细胞的清除,缓解神经元的进一步损伤,因此针对MerTK调控凋亡及氧化应激通路或可改善HIBD。本次实验研究中,我们显示OGD/R致小鼠HT22细胞活力显著降低,LDH漏出及细胞凋亡率明显增加的同时,HDAC11及MerTK表达升高,凋亡蛋白caspase-12和Bax显著上调。HDAC11特异性抑制能明显增强细胞活力、降低细胞凋亡率,显著下调caspase-12及Bax,而进一步上调MerTK表达。有报道,HIBD发生后,小胶质细胞通过TREM2上调吞噬功能介导突触丢失,但适当的吞噬作用有利于维持稳态[19]。已有研究表明,局灶性脑缺血后,小胶质细胞MerTK瞬时上调促进凋亡细胞的清除维持脑内稳态,而敲除MerTK导致凋亡碎片累积加重细胞损伤[20]。结合这些实验结果,我们认为,OGD/R致小鼠HT22细胞MerTK表达增加是一种保护性机制,而特异性抑制HDAC11可明显改善OGD/R致小鼠HT22细胞损伤,其机制可能通过进一步上调MerTK,促进凋亡细胞的清除,减轻凋亡细胞释放细胞因子引发更激烈的级联反应。
HIBD发生时,细胞内的活性自由基氧化生物分子破坏细胞脂质、蛋白质和核酸,并在HIBD发生后启动相关信号通路,导致细胞死亡和组织损伤[21]。MDA是脂质过氧化的终产物之一,SOD和GSH具有协同抗氧化作用。有研究表明MerTK通路的激活能促进小鼠原代巨噬细胞在氧化应激损伤中存活[22]。敲除HDAC11基因调控HO-1及Nrf-2改善果糖诱导的小鼠心肌细胞凋亡,减少活性氧产生,改善氧化应激损伤[23]。本次实验研究中,我们还发现,OGD/R致小鼠HT22细胞损伤、HDAC11及MerTK表达上调的同时,细胞MDA含量显著升高,SOD及GSH显著降低;抑制HDAC11显著降低OGD/R处理小鼠HT22细胞MDA含量,显著升高SOD及GSH水平。综合我们的实验结果及文献报道,抑制HDAC11明显改善OGD/R致小鼠HT22细胞损伤的机制,可能还涉及通过进一步升高MerTK水平,进而减轻氧化应激损伤。
总之,本实验研究表明,HDAC11上调MerTK表达,进而抑制细胞凋亡和氧化应激反应,参与OGD/R致小鼠HT22细胞损伤。但是需要进一步的体内外实验加以深入阐明,并明确HDAC11对MerTK是直接或间接调控的作用。
猜你喜欢
现代仪器与医疗(2021年6期)2022-01-18
中学生物学(2021年8期)2021-11-02
世界科学技术-中医药现代化(2020年2期)2020-07-25
食品工程(2020年3期)2020-01-05
中国果业信息(2019年1期)2019-01-05
中国畜牧兽医文摘(2018年6期)2018-07-28
计算机测量与控制(2017年6期)2017-07-01
西南军医(2016年6期)2016-01-23
决策与信息(2015年36期)2015-12-01
科学启蒙(2015年8期)2015-08-07
