
基于席夫碱配体构筑的LnⅢ2配合物的结构、荧光性质及生物活性
2023-02-03 10:22辛晓艳陈凤姣李文钰捷杨王文敏
无机化学学报 2023年1期
辛晓艳 陈凤姣 李文钰 王 捷杨 晨 李 敏 石 瑛*, 王文敏*,,
(1太原师范学院化学与材料学院,晋中 030619)
(2太原师范学院碳中和研究院,晋中 030619)
近年来,稀土配合物由于其新颖独特的结构,在磁性[1‑4]、发光[5‑7]、催化[8‑10]以及生物活性[11‑13]等领域存在潜在的应用价值而备受关注。在发光材料方面,一种优良的发光材料应在荧光分析中不仅具有高灵敏度,而且具有高选择性[14]。稀土元素中的EuⅢ和TbⅢ作为代替放射性同位素和非同位素标记的荧光探针具有很大的应用潜力。TbⅢ已被广泛用于研究DNA和生物体内镁离子的作用。稀土离子作为发光探针,不仅可用于研究生物分子和金属离子结合部位和结构类型[15],还可用来研究特定的物理化学条件下蛋白质具体的平衡构象。根据配合物的结构,一般可获得配合物的中心离子的格位数、中心离子的局部对称性、配位体形式电荷之和、直接与金属离子键合水的数目及2个金属离子间的距离等结构信息[16]。使用稀土离子作为生物分子的荧光探针,具有量子产率高、Stokes位移大、发射峰窄、激发和发射波长理想及荧光寿命长等优点[17]。此外,可以通过荧光探针分析方法对某些含有特殊结构的化合物进行特异性检测,因此荧光探针分析方法的应用相对于其他检测方法更为广泛[18]。
在生物活性方面,稀土配合物具有抑菌谱广以及杀菌能力强的优势,将其作为杀菌与消炎药物的效果较为显著。据大量实验证实,稀土金属离子与席夫碱组合而成的稀土配合物具有一定的生物活性,能抑制细菌生长、抗病毒以及抗肿瘤,由于协同效应,稀土配合物的生物活性比席夫碱化合物更强[19‑20]。2017 年 Yousif和 Majeed 等[21]研究了稀土席夫碱配合物的抗菌活性。研究表明,稀土席夫碱配合物具有良好的生物活性,特别是抑菌活性。席夫碱化合物具有丰富的配位原子和多种配位模式,因此稀土席夫碱配合物的合成比较灵活。近几年,关于稀土配合物在生命科学领域的研究不断深入,稀土配合物与DNA相互作用的研究有不少报道[22‑24]。DNA分子具有平行堆积的碱基聚合的阴离子磷酸骨架和2条核苷酸链螺旋形成的大沟和小沟,构成了金属配合物与DNA相互结合的位点,其结合方式可分为共价结合和非共价结合,非共价结合又可分为插入结合、沟面结合和静电结合。金属配合物与DNA相互作用研究成为当前生物无机化学领域的研究热点。
基于以上研究背景,我们探索了一种有效的合成策略,通过设计与合成具有丰富配位点(N、O原子)和多种配位模式的多齿席夫碱配体(E)‑N′‑(3‑乙氧基‑2‑羟基亚苄基)‑3‑羟基吡啶甲酰肼(H2L,图1),与稀土盐Ln(acac)3·2H2O(Ln=Tb、Ho、Er;acac-=乙酰丙酮根)反应,构筑了3例结构新颖的双核稀土配合物1~3。通过X射线单晶衍射、元素分析、热重分析和红外光谱等分析手段对配合物进行了表征。采用打孔抑菌圈法研究了配合物的抑菌活性。用荧光光谱仪测试了该类配合物的荧光发光性能。采用循环伏安(CV)法、紫外可见光谱法、琼脂糖凝胶电泳法和荧光光谱法研究了该类配合物与DNA相互作用机理,为该类配合物在抗菌和光致发光领域的研究和应用提供一定的依据。
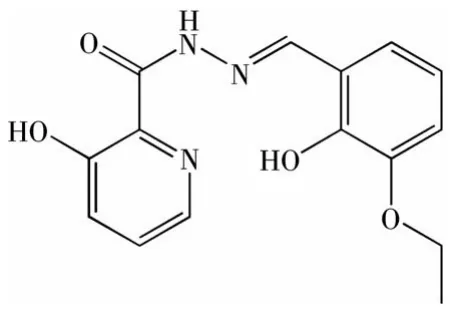
图1 配体H2L的结构Fig.1 Structure of H2L
1 实验部分
1.1 试剂和仪器
3‑乙氧基水杨醛、3‑羟基‑2‑吡啶甲酰肼、2,4‑戊二酮和六水合硝酸稀土盐(Ln(NO3)3·6H2O)购于安耐吉化学有限公司。常用溶剂(乙醇、乙腈、二氯甲烷、DMSO)购于科密欧试剂有限公司。大肠杆菌(E.coli)、枯草芽孢杆菌(B.subtilis)、金黄色葡萄球菌(S.aureus)、白色念珠菌(C.albicans)购于北京陆桥科技有限公司。营养琼脂培养基成分生化试剂(分析纯)购于北京奥博兴生物科技有限公司。三羟甲基氨基甲烷(Tris)、6X Glycerol Gel Loading Buffer、小牛胸腺DNA(CTDNA)和溴化乙锭(EB)购于生工生物工程(上海)股份有限公司。根据已报道的文献中的方法合成了多齿席夫碱配体H2L[25‑26]。按照文献[27]报道的方法合成了Ln(acac)3·2H2O(Ln=Tb、Ho、Er)。
配合物1~3的元素分析是在Perkin‑Elmer 2400分析仪上进行。用Bruker Tenor 27分光光度计测试红外光谱数据。粉末X射线衍射(PXRD)数据使用Rigaku Ultima Ⅳ仪器测试,采用CuKα射线为入射光源(λ=0.154 056 nm),扫描速度为 5(°)·min-1,扫描范围2θ=5°~50°,工作电压为40 kV,电流为25 mA。使用NETZSCHTG 409PC热重分析仪记录热重分析数据。紫外可见光谱测试在PERSEE TU‑1950型紫外可见分光光度计上进行。荧光光谱测试在港东科技的F‑320型荧光光谱仪上进行。生物活性试验采用YXQ‑LS‑30SII立式灭菌器、LRH‑150F生化培养箱、JB‑CJ‑1000FX超净工作台、辰华CHI750E电化学工作站。
1.2 配体H2L和配合物1~3的合成
称取 3‑羟基‑2‑吡啶甲酰肼(20.0 mmol,3.08 g)、3‑乙氧基水杨醛(20.0 mmol,3.32 g)于10 mL甲醇中,室温下搅拌4 h,抽滤收集白色沉淀物,用甲醇洗涤。粗品在真空环境下干燥24 h,得到配体H2L(图2)。元素分析按 C15H15N3O4的计算值(% ):C,59.74;H,4.97;N,13.93。实验值(% ):C,59.69;H,4.90;N,13.94。 IR(KBr,cm-1):3 287(m),3 072(w),2 947(w),2 931(w),2 891(w),2 362(m),1 661(s),1 627(m),1 611(m),1 551(m),1 532(m),1 489(m),1 446(s),1 415(m),1 354(w),1 337(m),1 278(w),1 247(s),1 205(s),1 187(w),1 137(w),1 095(w),1 072(m),949(w),910(w),874(w),817(w),751(s),711(w),643(w),533(w)(图S1,Supporting information)。1H NMR(400 MHz,CDCl3):δ11.61(s,1H),10.86(s,1H),8.43(s,1H),8.09(dd,J=4.2,1.5 Hz,1H),7.39(qd,J=8.5,2.8 Hz,2H),7.26(s,1H),7.06~6.75(m,3H),4.14(q,J=7.0 Hz,2H),1.48(t,J=7.0 Hz,3H)(图S2)。
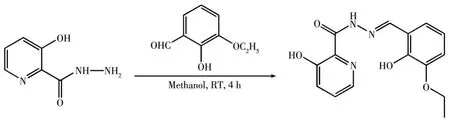
图2 配体H2L的合成路线Fig.2 Synthetic route of H2L
配合物1~3的合成方法相似,以下以1的合成为例说明。称取 Tb(acac)3·2H2O(0.025 mmol,0.021 g)、H2L(0.025 mmoI,0.008 g)加入到体积为20 mL的玻璃小瓶中,加入15 mL CH3OH/CH2Cl2/CH3CN(3∶1∶1,V/V)混合溶液后室温下搅拌30 min。在80℃下恒温反应24 h。然后冷却到室温,得到了适合单晶衍射的黄色块状晶体。配合物1~3的合成方法如图3所示。
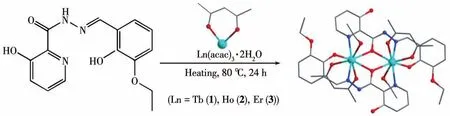
图3 配合物1~3的合成路线Fig.3 Synthetic routes of complexes 1‑3
[Tb2(acac)2(L)2(C2H5OH)2](1)的产率为 38% (基于Tb(acac)3·2H2O)。元素分析按 C44H52Tb2N6O14的计算值(% ):C,43.75;H,4.30;N,6.96。实验值(% ):C,43.76;H,4.29;N,6.95。IR(KBr,cm-1):3 427(m),2 983(w),2 953(w),2 927(w),2 897(w),2 874(w),2 365(w),1 600(s),1 568(m),1 551(m),1 537(m),1 459(s),1 406(m),1 383(w),1 362(w),1 343(w),1 287(s),1 248(s),1 223(m),1 197(s),1 164(w),1 076(s),1 029(w),992(w),943(w),914(w),883(w),860(w),833(w),797(w),748(s),721(w),665(w),615(w),586(m),540(m)(图S1)。
[Ho2(acac)2(L)2(C2H5OH)2](2)的产率为38% (基于Ho(acac)3·2H2O)。元素分析按C44H52Ho2N6O14的计算值(% ):C,43.32;H,4.26;N,6.89。实验值(% ):C,43.33;H,4.25;N,6.90。IR(KBr,cm-1):3 427(m),2 983(w),2 953(w),2 378(w),1 600(s),1 568(m),1 551(m),1 537(m),1 461(w),1 406(m),1 383(w),1 362(w),1 343(w),1 281(s),1 254(w),1 223(m),1 197(s),1 164(w),1 155(m),1 135(w),1 082(s),1 029(w),943(w),897(w),883(w),860(w),833(w),797(w),745(w),721(w),665(w),615(w),586(m),543(m)(图 S1)。
[Er2(acac)2(L)2(C2H5OH)2](3)的产率为 39% (基于Er(acac)3·2H2O)。元素分析按 C44H52Er2N6O14的计算值(% ):C,43.15;H,4.25;N,6.86。实验值(% ):C,43.14;H,4.26;N,6.87。IR(KBr,cm-1):3 427(m),2 983(w),2 953(w),2 378(w),1 600(s),1 568(m),1 551(m),1 537(m),1 461(w),1 406(m),1 383(w),1 362(w),1 343(w),1 281(s),1 254(w),1 223(m),1 197(s),1 164(w),1 118(s),1 029(w),992(w),943(w),914(w),883(w),860(w),833(w),797(w),745(s),721(w),665(w),619(s),583(m),543(m)(图 S1)。
1.3 配合物1~3晶体结构的测定
选取形状规则、大小合适的配合物1~3的固态单晶样品,其大小为0.39 mm×0.26 mm×0.16 mm(1)、0.36 mm×0.25 mm×0.13 mm(2)和0.36 mm×0.26 mm×0.21 mm(3)。将单晶样品放在CCD单晶衍射仪上测定并收集晶体衍射数据,测试配合物1和配合物3单晶采用MoKα射线(λ=0.071 073 nm)为入射光源,配合物2单晶采用CuKα射线(λ=0.154 178 nm)为入射光源,在150 K时,以ω‑φ的扫描方式收集衍射点。晶体结构用SHELXS‑97[28]中的直接法解出,并用SHELXL‑97[29]对结构中的非氢原子坐标及其各向异性热参数进行基于F2的全矩阵最小二乘法修正至收敛。所有氢原子均为理论加氢并按跨式模型(riding model)精修,其Uij为其母原子Uij的 1.2 倍~1.5倍。由上述方法得到的配合物1~3的主要晶体学数据列于表1,重要的键长和键角见表S1~S3。
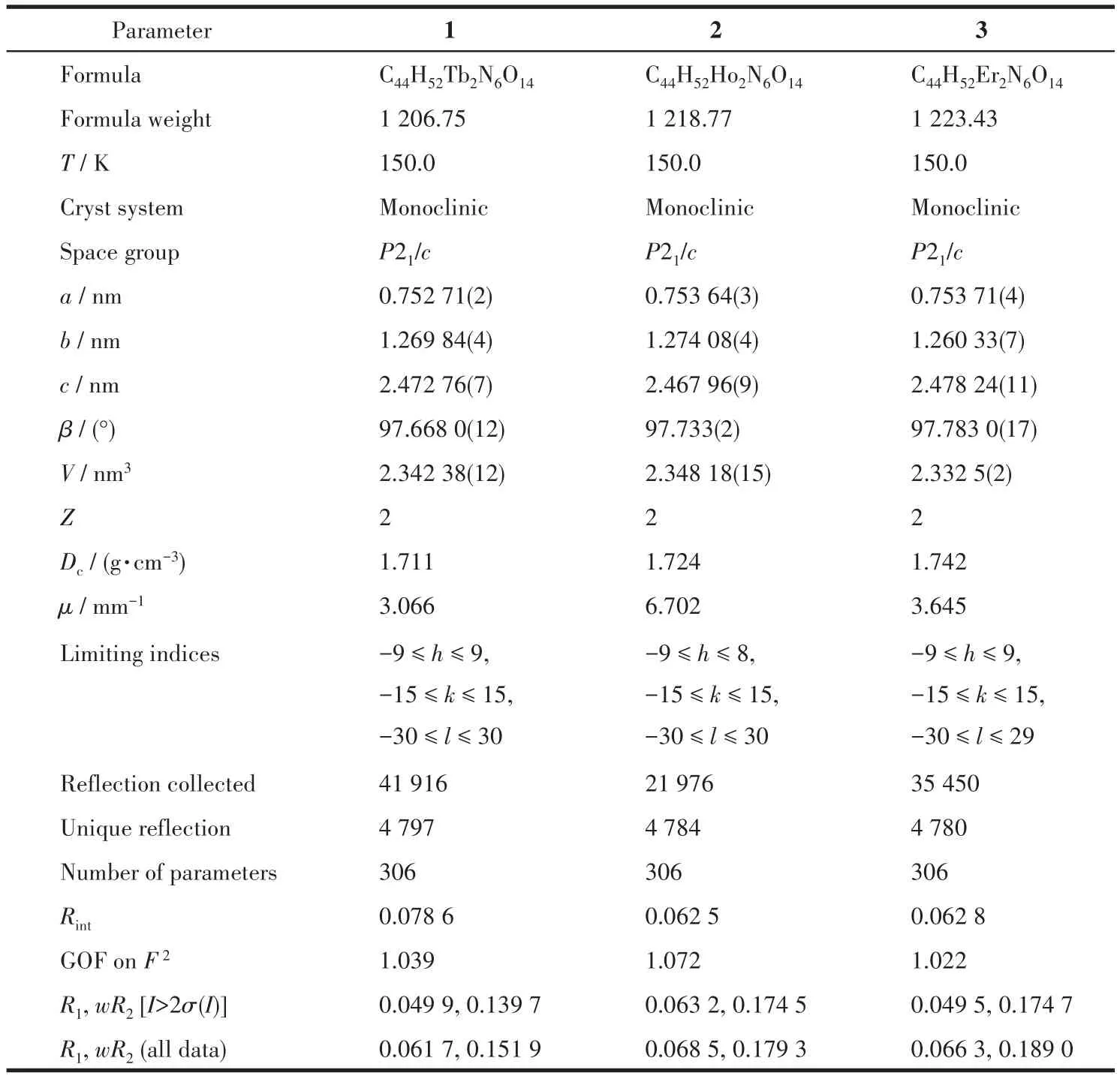
表1 配合物1~3的晶体学数据和精修参数Table 1 Crystallographic data and structure refinements for complexes 1‑3
CCDC:2183865,1;2183867,2;2183869,3。
1.4 生物活性测定
1.4.1 抑菌圈试验
将配合物1~3用DMSO分别配成溶液,采用打孔抑菌圈法[30‑31]测定了配合物对4种细菌(E.coli、B.subtilis、S.aureus、C.albicans)的抑菌活性。将活化好的细菌稀释成不同浓度的菌液,移取100 μL稀释后的各不同浓度的菌液,在LB固体培养基上涂布均匀,放置在37℃的生化培养箱中培养10 h左右,最终确定合适的菌液浓度为8×104CFU·mL-1,用来测试抑菌圈。用已灭菌的4 mm不锈钢管在平板上打孔,孔内注入60 μL抗菌剂(0.01 g·mL-1),4 ℃下预扩散2 h,在37℃的生化培养箱中培养10 h左右,用游标卡尺测量抑菌圈直径大小。
采用微量液体培养基倍比稀释法测定配合物对不同菌株的最小抑菌浓度(minimal inhibitory cconcentration,MIC)值(4种供试细菌都用LB液体培养基培养)。将不同浓度的抑菌剂混合液溶解于LB液体培养基中培养,然后接种细菌,通过细菌的生长与否,确定抑菌剂抑制受试菌生长的最低浓度,即 MIC 值[32‑37]。
1.4.2 配合物1~3与DNA相互作用
1.4.2.1 紫外可见光谱法
称 取 Tris(0.302 5 g)、HCl(6 mol·L-1,0.55 mL)、NaCl(0.146 3 g),加蒸馏水配制 Tris‑HCl/NaCl(500 mL,5 mmol·L-1)缓冲溶液,调节pH=7.25。取1.1 mL CTDNA 母液(2 μg·mL-1),用紫外可见光谱测定其A260/A280,使其在 1.8~2.0 之间。以 Tris‑HCl/NaCl(5 mmol·L-1)缓冲液(pH=7.25)为参比溶液,以 CTDNA溶液为空白对照。将配合物1~3用Tris‑HCl/NaCl缓冲溶液稀释,浓度为 0.45、0.22、0.11、0.055、0.028、0.014 mmol·L-1。 将 10 μL 配 合 物 加 入 到 3 mL CTDNA溶液中,在190~500 nm处测定其紫外可见吸收光谱。
1.4.2.2 CV法
CV法测试中,工作电极为玻碳电极,参比电极为饱和甘汞电极,对电极为铂丝电极。扫描电位范围为-0.15~0.8 V,扫描速度为0.1 V·s-1,采样间隔为0.001 V,静置时间为2 s,样品浓度为0.45 mmol·L-1,DNA的质量浓度为2 μg·mL-1。
1.4.2.3 凝胶电泳法
向1.5 mL Eppendorf管中分别加入1 μL的不同浓度梯度的配合物溶液(0.4、0.2、0.1、0.05、0.025 mmol·L-1)、2 μL的pBR322DNA 溶液(0.5 mg·mL-1)、7 μL的Tris‑HCl/NaCl反应缓冲溶液(5 mmol·L-1,pH=7.25),使反应体系总体积保持10 μL,高速离心混合均匀,放置于37℃恒温槽避光反应20 min。随后恒温反应20 min,加入6X Glycerol Gel Loading Buffer终止反应,琼脂糖凝胶电泳20 min后分析结果。
1.4.2.4 荧光光谱法
荧光光谱法测试中,CTDNA和EB的质量浓度分别稳定在 2 μg·mL-1和 0.06 mg·mL-1。按体积比1∶1充分混合,避光放置1 h。向样品池中加入3 mL EB‑CTDNA混合溶液,在285 nm激发波长下,在540~700 nm范围内测量其荧光发射光谱。然后,将配合物溶液(0.45 mmol·L-1)每次滴加1 μL 到EB‑CTDNA体系中,滴入后混合均匀,室温孵育1 min,测量其荧光发射光谱。
2 结果与讨论
2.1 配合物1~3晶体结构分析
单晶X射线衍射数据表明,配合物1~3是同构的,因此以配合物1为例进行具体的晶体结构描述(图4)。1属于单斜晶系P21/c空间群,其结构主要由2个TbⅢ离子、2个乙酰丙酮根(acac-)、2个L2-及2个C2H5OH组成。中心的Tb1离子是八配位(图5a),与Tb1离子配位的6个氧原子来自2个L2-的3个氧原子(O3、O2和O2a)、1个乙醇分子的O7氧原子和1个acac-的2个氧原子(O5和O6),2个氮原子来自2个L2-(N1和N3)。如图5b所示,八配位的TbⅢ离子呈三角形十二面体配位构型,根据SHAPE 2.0软件计算得出(表S4)。L2-和acac-的配位模式如图6所示,L2-通过多齿螯合方式包裹2个中心TbⅢ离子,acac-通过双齿螯合方式连接每个中心TbⅢ离子。2个中心TbⅢ离子通过2个L2-的2个μ2‑O(O2和O2a)桥联形成一个平行四边形的Tb2O2核心。在Tb2O2核心结构中,Tb1…Tb1a间的距离为 0.397 11(6)nm,Tb1—O2—Tb1a和 O2—Tb1—O2a的键角分别是112.03(17)°和 67.97(17)°,在 1 中,Tb—O 的键长为0.2357(5)~0.2391(5)nm,Tb—N的键长为0.2485(6)~0.256 3(6)nm。 O—Tb—O 键角在 67.97(17)°~146.28(16)°范围内,O—Tb—N 键角在 63.63(17)°~113.32(17)°范围内,所有键长和键角都在正常范围内,与文献报道的双核Tb的键角一致[38‑40]。
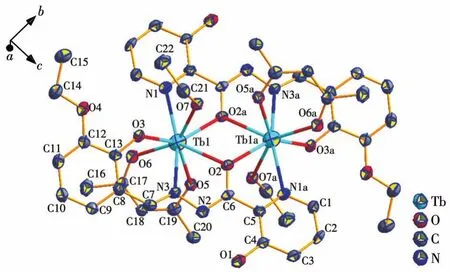
图4 配合物1的椭球率50% 的晶体结构图Fig.4 Molecular structure of complex 1 shown with 50% probability displacement ellipsoids
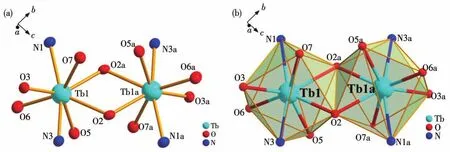
图5 配合物1中Tb2的配位环境(a)和Tb2离子的几何多面体(b)Fig.5 Coordination environment of Tb2(a)and geometric polyhedron of Tb2ions(b)in complex 1
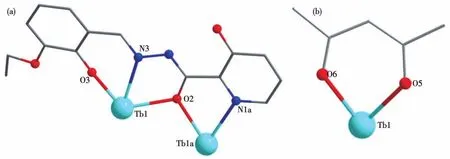
图6 L2-(a)和acac-(b)的配位模式Fig.6 Coordination modes of L2-(a)and acac-(b)
2.2 配合物1~3的PXRD和热重分析
为了检验配合物1~3的相纯度,在室温下对3个配合物的单晶样品分别进行了PXRD测试。将测试得到的PXRD数据与通过单晶结构模拟计算的理论值进行了对比,如图S3所示,实验数据图与拟合的PXRD图主要峰的位置和形状基本吻合,表明收集的1~3的晶体样品具有较高的相纯度。
为了进一步探究配合物1~3的热稳定性,在N2保护下,对它们的晶体粉末进行热重测试(温度范围20~800℃,升温速率10℃·min-1),其热重曲线如图S4所示。随着温度的升高,1~3的失重过程相似,故仅对3进行详细描述。在80℃之前3可以稳定存在,80~207℃之间失重7.31%,对应失去2个配位的乙醇分子(理论值为7.53% )。207~350℃之间失重16.43%,对应失去2个配位的辅助配体acac-离子(理论值为16.36% )。随后,3的骨架在350~800℃的温度范围内逐渐分解。
2.3 配合物1~3的紫外可见吸收光谱
室温下,在甲醇溶液中获得了多齿席夫碱配体H2L、Tb(acac)3·2H2O和配合物1~3的紫外可见光谱(图S5)。可以清楚地观察到H2L分别在206、236和323 nm处有吸收带,归属于芳香环和C=N的n→π*和π→π*跃迁。Tb(acac)3·2H2O 在 202、241和290 nm处出现3个吸收带,这是acac-中羰基(C=O)的π→π*跃迁引起的。1~3表现出相似的吸收带,可以观察到在336 nm处的吸收带归属于席夫碱配体L2-,在202和239 nm处的吸收带,应归属于acac-辅助配体。此外对比1~3与H2L的紫外可见谱图可见,300~338 nm处的吸收带发生了红移,这可能是由于H2L与稀土离子的配位效应。
2.4 荧光性质
如图7所示,配体H2L用260 nm波长激发时,在518 nm处有特征发射峰。在296 nm波长激发下,配合物1的发射光谱表现出中心离子TbⅢ的特征发射。1在490、545、585和620 nm处观察到4个特征发射峰,对应 TbⅢ离子5D4→7FJ(J=6、5、4、3)的跃迁。1在乙腈溶液中测得的荧光寿命是139 μs(图S6)。在260 nm波长激发下,配合物2和3分别在532和530 nm处观察到一个特征发射峰,均可归因于配体H2L的荧光发射。2和3的发光均为配体发光,但最强发射峰发生红移且强度增大,说明中心离子对配体的发光有敏化作用。

图7 室温下配合物1~3和配体H2L的固体荧光谱图Fig.7 Solid‑state fluorescent spectra of complexes 1‑3 and ligand H2L at room temperature
2.5 抑菌活性分析
根据文献[41‑43]报道的方法,采用打孔抑菌圈法和MIC法测定了配合物、配体H2L以及稀土离子的抗菌活性。图S7是配合物1~3对E.coli、B.subtilis、S.aureus、C.albicans的抑菌圈图。图8数据显示,DMSO对4种细菌的抑菌圈小于7 mm,没有抑菌作用;稀土离子对E.coli的抑菌作用略强于其他3种细菌;H2L和1~3对4种细菌具有良好的抑菌作用,尤其对C.albicans的抑菌作用略强于其他3种细菌。1~3对4种细菌的抑菌圈均大于20 mm,表明1~3的抑菌作用强于单一配体或稀土离子,这可能是配体和稀土离子之间的协同作用导致的[44]。用MIC法测定的抑菌试验结果如表S5所示。结果表明,1~3对4种测试菌的MIC低于50 μg·mL-1,这也说明1~3的抑菌效果强于配体和稀土离子。
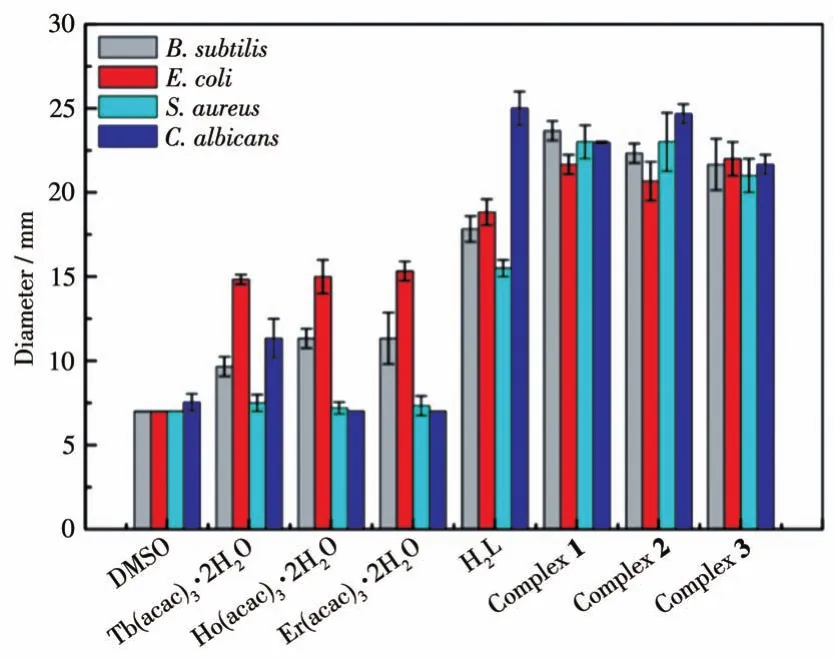
图8 配合物1~3、配体H2L及Ln(acac)3·2H2O(Ln=Tb、Ho、Er)的抑菌活性圈直径Fig.8 Antibacterial activity circle diameters of complexes 1‑3,ligand H2L,and Ln(acac)3·2H2O(Ln=Tb,Ho,Er)
2.6 配合物1~3与DNA相互作用
2.6.1 紫外可见光谱法分析
紫外可见吸收光谱法是研究小分子与核酸相互作用机制的一种简单而常用的方法[45‑48]。由于DNA分子中嘌呤碱基和嘧啶碱基的共轭双键的影响,DNA分子在240~290 nm处会有较强的紫外吸收带,最大吸收峰在260 nm左右,而金属配合物也有吸收带,因此,可以根据配合物与DNA分子相互作用前后谱带的变化来判断配合物的作用方式。从图9可以看出,1~3与CTDNA相互作用后,配合物‑CTDNA复合物体系的紫外吸收光谱比单独的CTDNA有明显的红移,随着浓度的增大,红移幅度也增大,而且在260 nm处发生减色效应,2和3的减色效应最明显。由这种现象可以推断1~3与DNA发生插入结合。
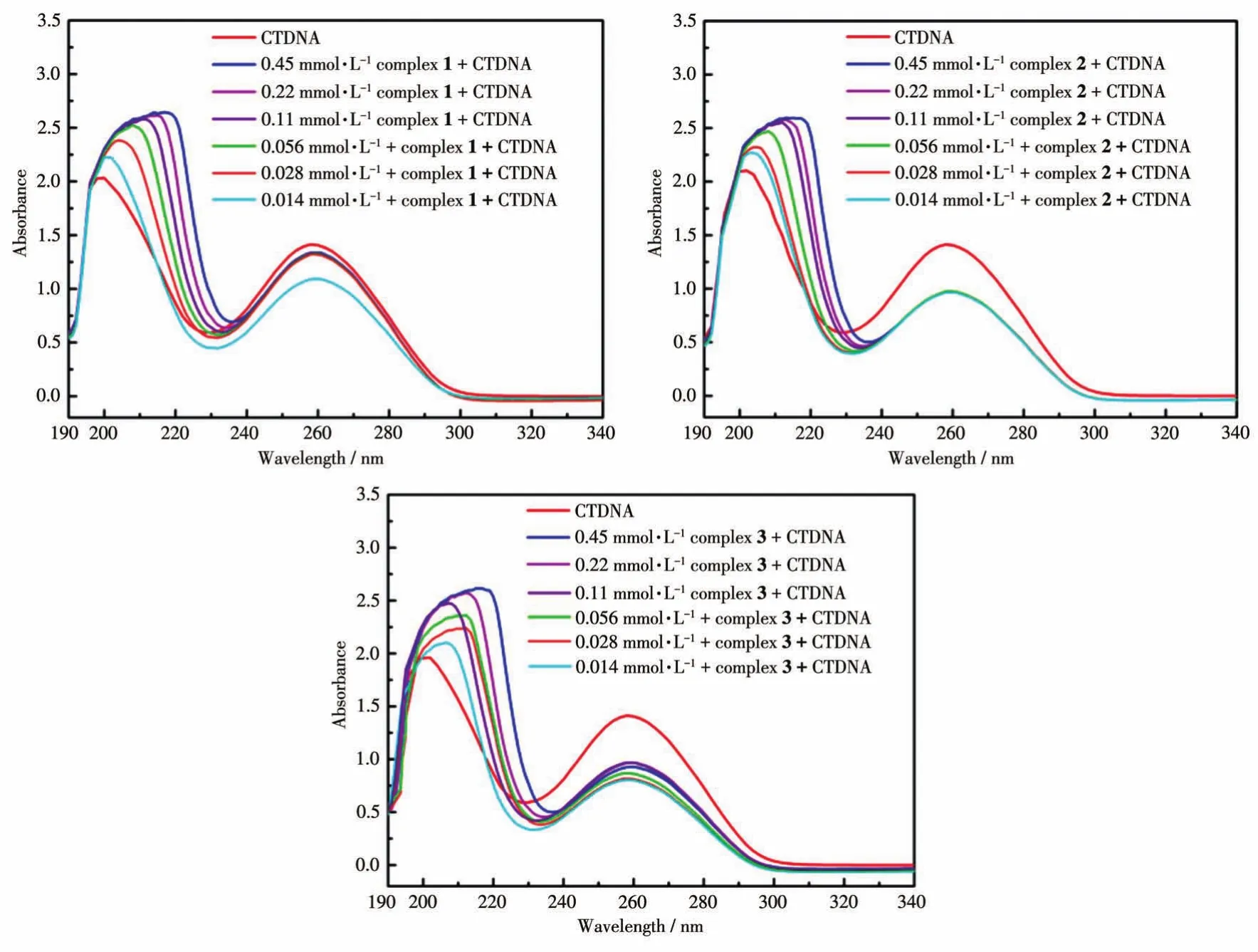
图9 配合物1~3与CTDNA相互作用的紫外光谱图Fig.9 UV Spectra for the interaction between complexes 1‑3 and CTDNA
2.6.2 CV法分析
根据加入DNA前后配合物电化学性质的变化,可以判断配合物与DNA的相互作用模式。当配合物分子与DNA发生插入结合时,配合物的扩散系数减小,导致其还原峰电流减小,配合物分子的CV曲线式量电位会正移;当配合物与DNA中带负电的磷酸基团静电结合时,配合物分子的CV曲线式量电位会负移[49‑51]。从图10可以看出,配合物1~3的CV曲线式量电位正移,其中3的正移最明显,这表明1~3与DNA可能发生了插入结合[52‑54]。
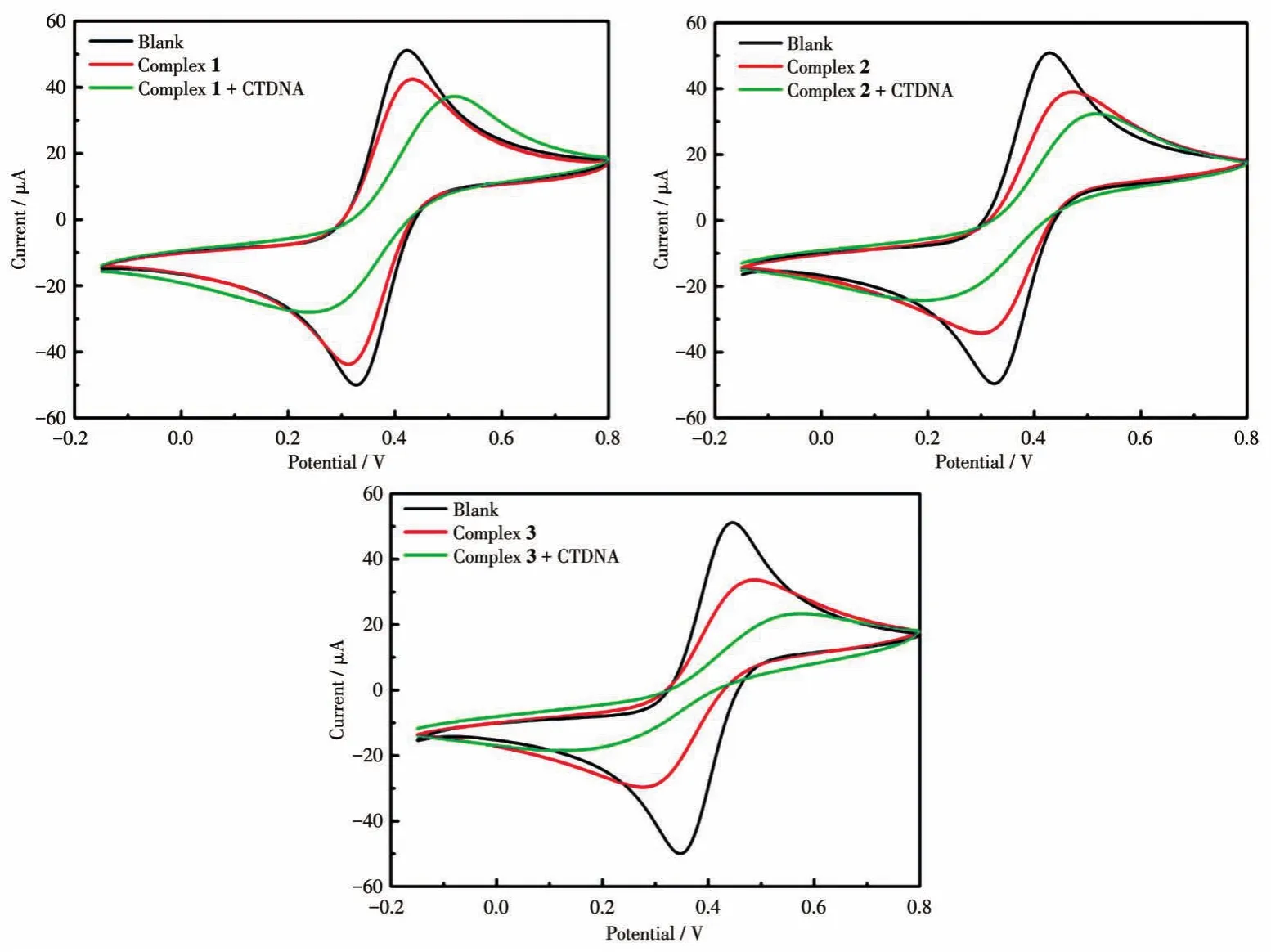
图10 配合物1~3与DNA相互作用的CV曲线Fig.10 CV curves for the interaction between complexes 1‑3 and CTDNA
2.6.3 凝胶电泳法分析
当配合物与超螺旋DNA相互作用时,共价的闭合DNA双螺旋将被打开,DNA分子被切割成多个小分子DNA。凝胶电泳法可以揭示配合物引起的DNA分子结构的变化。因为配合物1~3是同构的,我们选取2、3作为代表分析它们与pBR322DNA之间的相互作用。如图S8所示,当配合物与pBR322DNA反应时,超螺旋pBRDNA条带的亮度明显比空白pBR322DNA低,可以很明显看到pBR322DNA切割出另一条条带,这表明超螺旋pBRDNA条带的数量减少,并被切割成几条相对分子质量较低的DNA条带。因此,配合物2、3与pBRDNA相互作用较强,与pBRDNA发生了较强的切割作用。
2.6.4 荧光光谱法分析
已知EB与DNA结合以后能发射荧光,可利用其与DNA的结合研究配合物与DNA之间作用机制。EB本身发射的荧光强度弱且与DNA的结合能力较强,配合物与DNA分子结构发生特异性结合会引起荧光的部分猝灭现象[55‑59]。由荧光光谱法实验可以计算配合物与DNA的结合常数进而判断二者间的作用力大小。根据Stern‑Volmer方程[60]:I0/I=1+Ksqr,其中I0为未加配合物时EB‑DNA体系的荧光强度,I为加入不同浓度的配合物后EB‑DNA复合体系的荧光强度,r为配合物与DNA浓度之比,Ksq是猝灭常数,当I0/I与r呈线性关系,Ksq可以由直线斜率求出,该常数可用于比较配合物分子与DNA的结合能力。图11为配体H2L和3个配合物与EB‑DNA复合体系的荧光猝灭图,由Stern‑Volmer方程拟合结果得到H2L和配合物1~3的Ksq分别为0.08、0.82、1.28和0.77,说明1~3与DNA的作用强于配体H2L,配合物分子与DNA发生了插入结合。

图11 配体H2L和配合物1~3对EB‑DNA复合体系的荧光猝灭Fig.11 Fluorescence quenching of EB‑DNA complex system by ligand H2L and complexes 1‑3
3 结论
以多齿席夫碱H2L为配体与Ln(acac)3·2H2O(Ln=Tb、Ho、Er)反应,设计、合成了3例新的双核Ln配合物[Ln2(acac)2(L)2(C2H5OH)2](Ln=Tb(1)、Ho(2)、Er(3))。单晶X射线衍射结构表明:中心LnⅢ离子通过2个μ2‑O原子相互连接,形成一个平行四边形的Ln2O2核心。采用抑菌圈法和最小抑菌浓度法研究了配合物1~3对枯草芽孢杆菌、大肠杆菌、金黄色葡萄球菌和白色念珠菌的抑制作用。结果表明,配合物的抑菌作用强于单一配体或稀土离子。用循环伏安法、紫外光谱法和荧光光谱法研究了配合物1~3与DNA的相互作用机理。研究结果对设计、合成新的抗菌金属配合物药物具有一定意义。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
化学与粘合(2020年4期)2020-09-11
当代陕西(2019年6期)2019-04-17
分析化学(2018年12期)2018-01-22
凿岩机械气动工具(2017年1期)2017-05-17
中国塑料(2016年10期)2016-06-27
中成药(2016年8期)2016-05-17
铜业工程(2015年4期)2015-12-29
应用化工(2015年7期)2015-12-24
外语学刊(2014年3期)2014-12-03
