
N缺陷g‑C3N5修饰S掺杂苝酰亚胺增强可见光自芬顿苯酚氧化耦合Cr还原
2023-02-03 10:22:56徐凯旋亢玉龙贺红斌高晓明赵晨宇任瑞阳
无机化学学报 2023年1期
徐凯旋 亢玉龙 贺红斌 高晓明 赵晨宇 任瑞阳
(延安大学化学与化工学院,陕北低阶煤清洁高效利用协同创新中心,陕西省化学反应工程重点实验室,延安 716000)
0 引言
传统化石能源的消费引起环境污染和能源短缺等问题,成为人类可持续发展面临的重大挑战。因此,在当前“双碳目标”“能耗双控”背景下,探索新型清洁能源与绿色污染治理技术引起了人们的广泛关注。以太阳能驱动的光催化技术是一种理想的能源利用及污染治理的途径,为开发新型能源、解决环境污染提供了新的契机。光催化技术所面临的核心问题是寻找性能优良的光催化剂,因此高效光催化剂的筛选与制备是光催化研究的核心课题。近年来,如g‑C3N4[1‑2]、聚苯 胺[3‑4]、苝 酰亚胺(PDI)以及对应的衍生物等[5‑6]是以资源丰富的 C、H、O、N构建的有机半导体,可通过π‑π作用和氢键精确设计,分子结构易于调控,结构多样;同时,有机半导体电子结构容易调控,价带电位较深,使其具有很强的氧化能力。PDI是一种应用较为广泛的n型有机超分子半导体[5],吸光范围宽,吸收边带可延伸到800 nm,因此具有优异的可见光催化性能。g‑C3N5是一种富氮n型无机半导体,在可见光范围有较高的富电子位,光响应范围宽、带隙窄(大约在2.2 eV左右)[7]。然而,在应用过程中它们也呈现出一个不可避免的缺点:光催化过程中光生电子-空穴容易复合,分离效率低,这对光催化效果具有不可忽视的影响。
通过将N缺陷的g‑C3N5(NVs)与有机半导体PDI结合,构建电子空穴传输距离长的异质结,使电子空穴发生空间分离,分别利用价带空穴的氧化能力和导带电子的还原能力,实现电子-空穴的空间分离,可以强化氧化半反应和还原半反应。因此,利用空间分离的电子-空穴,有可能在还原去除Cr时协同氧化降解苯酚。同时,Cr可以成为光自芬顿反应电子的转移位点,与光照产生的电子、H2O2形成一个光自芬顿反应过程,促进苯酚的降解与Cr的还原。基于此,本研究通过静电自组装制备了有机复合半导体NVs修饰S掺杂PDI(NVs/S‑PDI)异质结,并对其物相结构、表面化学组成、微观形貌、光吸收性能、光电化学性质等进行了研究。通过研究NVs/S‑PDI异质结对苯酚和Cr混合溶液的光催化氧化还原效果,考察价带空穴的氧化能力和导带电子的还原能力对苯酚和Cr的协同去除率。通过分析反应体系的H2O2产量、活性自由基种类等,讨论光催化效果提高原因和光催化机理。
1 实验部分
1.1 试剂
主要试剂有苝‑3,4,9,10‑四羧酸二酐(C24H8O6,PDA)、硫代乙酰胺(CH3CSNH2)、三氨基‑1,2,4三唑(C2H4N4)、乙酸钠(CH3COONa)、咪唑(C3H4N2)、三乙胺((C2H5)3N)、对苯醌(BQ)、异丙醇(IPA)、乙二胺四乙酸二钠(EDTA‑2Na)、重铬酸钾(K2Cr2O7)。
1.2 样品制备
1.2.1 NVs的制备
将2 g C2H4N4、10 g KBr溶解于20 mL去离子水中,在70℃油浴中搅拌8 h。将所得固体研磨成粉,在550℃马弗炉中保持4 h。之后用去离子水洗涤数次,65 ℃真空干燥 12 h,获得 g‑C3N5。将 0.2 g g‑C3N5在550℃管式炉中Ar气氛下煅烧2 h,自然冷却至室温,得到NVs[8]。
1.2.2 S‑PDI的制备
将 3 mmol PDA、6 mmol CH3CSNH2、6 mmol CH3COONa、10 g C3H4N2加入水热釜,在120 ℃下保持5 h。冷却至室温,加入200 mL HCl(2 mol·L-1),室温下搅拌12 h。将过滤得到的固体用去离子水和无水乙醇洗涤多次,放入真空干燥箱在65℃下干燥,研磨成粉,得到PDI前体,记为PDI(0),其产率大约为15%。
将0.027 6 g PDI前体分散到15 mL去离子水中,超声30 min。加入41 μL三乙胺,搅拌10 min,超声30 min,置于60℃油浴中,加入1.4 mL硝酸溶液(4 mol·L-1),搅拌1 h。冷却至室温,用去离子水洗涤至中性,放入65℃真空干燥箱干燥12 h,得到S‑PDI,产率大约为91%。
1.2.3 NVs/S‑PDI的制备
称取0.027 6 g PDI,分散到15 mL去离子水中,超声30 min。加入41 μL(C2H5)3N,搅拌10 min,向其中加入一定量的NVs,超声30 min。置于60℃油浴中,加入1.4 mL 4 mol·L-1的硝酸溶液,继续搅拌1 h。冷却至室温,用去离子水洗涤至中性,放入65℃真空干燥箱干燥12 h,得到NVs/S‑PDI,记为x% NVs/S‑PDI(NVs的质量分数x% =10% 、20% 、30% 、50% )。制备过程如图1所示。
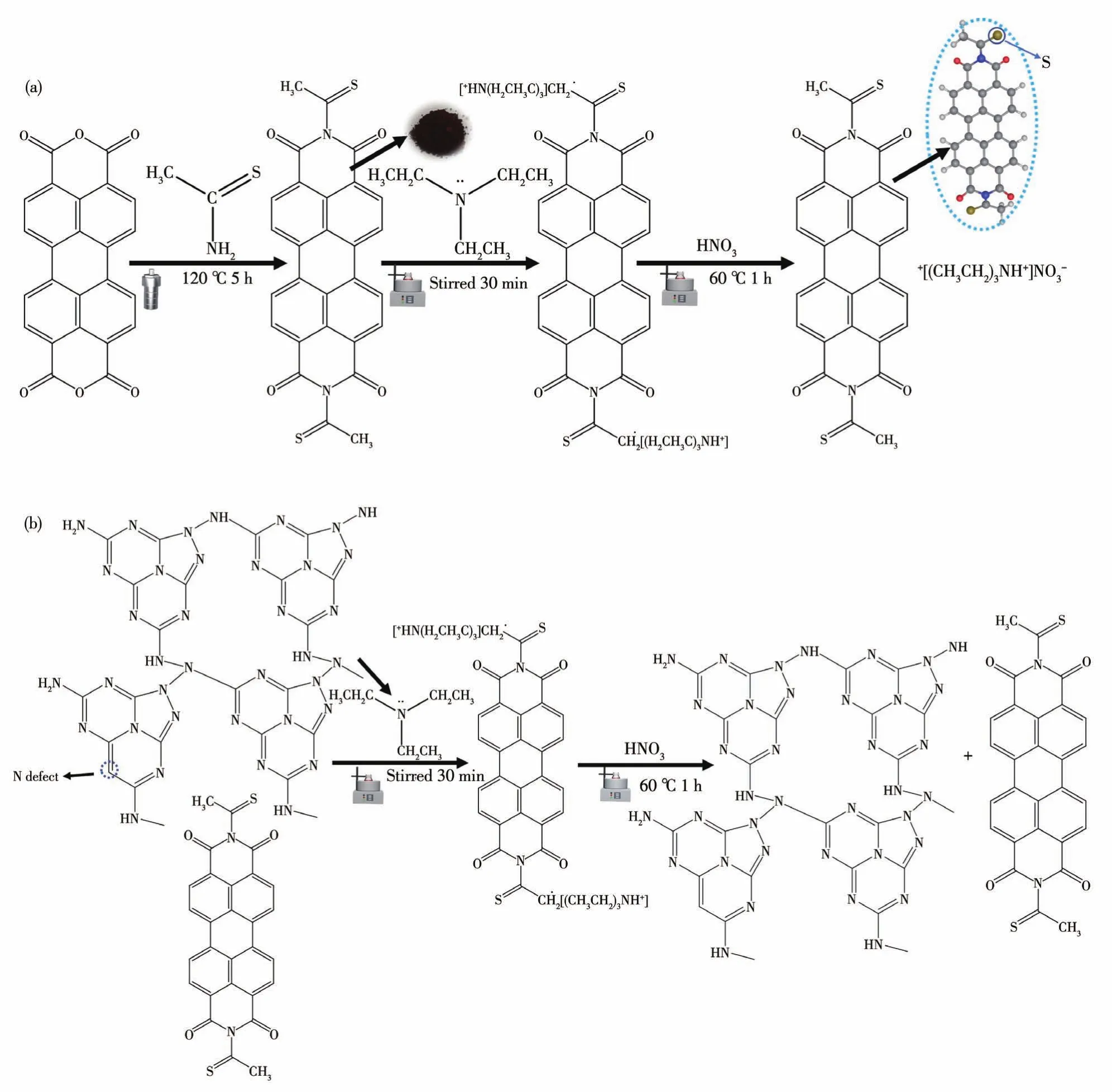
图1 (a)S‑PDI和(b)NVs/S‑PDI的制备过程Fig.1 Preparation process of(a)S‑PDI and(b)NVs/S‑PDI
1.3 光催化性能测试
将25 mg催化剂分散在25 mL 10 mg·L-1苯酚溶液(重铬酸钾溶液或苯酚与重铬酸钾混合溶液),避光暗反应0.5 h,使催化剂与反应物分子达到吸附-脱附平衡。之后用300 W氙灯光照3 h,隔一定时间取样分析。用紫外可见分光光度计在λ=352 nm处测定吸光度以确定Cr浓度,使用高效液相色谱在保留时间(tD)为5.75 min处测定苯酚浓度。产生的H2O2用碘量法测定。
1.4 表征方法
使用日本岛津的XRD‑7000型粉末X射线衍射仪(PXRD)测定催化剂的物相结构,CuKα射线,Ni滤光片,40 kV管电压,30 mA管电流,λ=0.154 18 nm,扫描范围20°~80°。采用日本电子的JSM‑6700型扫描电子显微镜(SEM,工作电压和电流分别为10 kV和10 μA)、JEM‑2100透射电子显微镜(TEM,加速电压为200kV)分析催化剂的微观形貌。采用日本岛津的IRAFFINITY‑1S型傅里叶变换红外仪分析催化剂的表面官能团,扫描范围为400~4 000 cm-1。使用美国PE的PHI‑5400 X射线光电子能谱仪(XPS)测定样品的元素组成和化学状态,AlKα单色化X射线源。采用日本岛津的UV‑2550型紫外可见分光光度计测定催化剂的紫外可见漫反射光谱(UV‑Vis DRS),BaSO4为参照物。采用日本日立的F‑4500荧光分析仪测定样品的光致发光光谱,激发波长为350 nm。采用上海辰华的CH1660D型电化学工作站分析催化剂的光电化学性质,负载催化剂的ITO玻璃为工作电极,Pt电极为对电极,Ag/AgCl电极为参比电极,0.5 mol·L-1Na2SO4溶液为电解液,300 W氙灯(λ≥400 nm)。采用 Malvern zetasizer nano series测试催化剂的ζ电位(pH=2~3)。
1.5 活性物质测试
在捕获活性物质的实验中,分别用BQ、IPA和EDTA‑2Na捕获·O2-、·OH 和 h+。其中 BQ、IPA、EDTA‑2Na的浓度分别为0.1、10和10 mmol·L-1。反应一定时间后,取2 mL溶液,0.22 μm滤膜过滤,使用高效液相色谱测定苯酚浓度。以可见光作为光源,用DMPO(5,5′‑二甲基‑1‑吡咯烷‑N‑氧化物)进行电子自旋共振(ESR)测试,在黑暗中测试1次,光照中测试2次,每次间隔4 min。
2 结果与讨论
2.1 样品的表征
图2为催化剂的XRD图。由图2a可知,g‑C3N5在 2θ=13.0°、28.3°处出现特征衍射峰,分别对应于(100)、(002)晶面[9]。通过 Ar气氛煅烧在 g‑C3N5表面引入N缺陷后,(002)晶面对应的衍射峰(2θ=27.9°)向高角度偏移,使得晶面间距减小,有利于电子的转移。PDI(0)、S‑PDI在 2θ=12.8°、17.38°、23.02°、25.19°、27.50°处出现特征衍射峰。S‑PDI的衍射峰基本没有变化,表明在自组装过程中,PDI的晶格结构没有发生改变。此外,与报道的U‑PDI[10]相比,S‑PDI的衍射峰发生了微小偏移,可能是由PDI分子中的CH3、S=C导致π‑π堆叠不对称性引起的。随着x% NVs/S‑PDI中NVs量的增加,2θ=12.8°、17.38°、23.02°、25.19°的特征衍射峰变弱。此外,由于分子间的π‑π堆叠的相互作用,相比于S‑PDI、NVs,x% NVs/S‑PDI在2θ=27.50°的特征衍射峰都产生了微小偏移(图2b)。这将导致S‑PDI的晶格界面间距减小,有利于电子传输。同时,x% NVs/S‑PDI在2θ=27.50°的特征衍射峰峰面宽度增大,可能是由于S‑PDI和NVs发生簇集,导致多个晶面被检测。x% NVs/S‑PDI在2θ=35.20°、43.40°的特征衍射峰峰强增大,表明S‑PDI和NVs存在强的相互作用[11],这种相互作用可能是NVs层与S‑PDI层间叠加导致的,使得特征衍射峰增强[12]。图2c为催化剂的FT‑IR谱图。从图中可知,1 200~1 600 cm-1范围的特征吸收峰属于NVs单元的C—N/C=N官能团。2 160 cm-1处的特征吸收峰属于 NVs单元的—C≡N 官能团[13‑14]。1 595 cm-1处的特征吸收峰归属于S‑PDI的芳香环官能团,1 655 cm-1处的特征吸收峰对应S‑PDI的C=S官能团,1 690 cm-1处的特征吸收峰则归于S‑PDI的C—O官能团[15],表明S‑PDI存在苯环和酰胺基团。相对于NVs和S‑PDI,30% NVs/S‑PDI的特征吸收峰的强度都有细微的降低,表明成功构建了NVs/S‑PDI异质结,且S‑PDI和 NVs存在强的相互作用[7]。S‑PDI、NVs的ζ电位测试结果如图S1a、S1b(Support‑ing information)所示,S‑PDI的ζ电位值为-38.3 eV,NVs的ζ电位值为57.1 eV,表明二者是通过静电自组装结合在一起。

图2 (a)所制备样品的XRD图;(b)所制备样品在20°~50°范围的XRD图;(c)NVs、S‑PDI和30% NVs/S‑PDI的FT‑IR谱图Fig.2 (a)XRD patterns of as‑prepared samples;(b)XRD pattern of as‑prepared samples in a range of 20°‑50°;(c)FT‑IR spectra of NVs,S‑PDI,and 30% NVs/S‑PDI
NVs中的氮缺陷可以用电子顺磁性共振(EPR)表征,结果如图3a所示。可以看出,g‑C3N5在g=2.003的特征峰强度较弱,而NVs在g=2.003的特征峰强度增强。因此,通过在Ar氛围下煅烧,得到了丰富N缺陷的NVs。由30% NVs/S‑PDI的XPS全谱图(图3b)可知,30% NVs/S‑PDI由C、N、O、S四种元素组成。NVs、S‑PDI、30% NVs/S‑PDI的C1s谱图如图 3c 所示,在 284.71 eV(上图)、284.77 eV(下图)、284.76 eV(中图)处的峰分别归属于 NVs、S‑PDI、30% NVs/S‑PDI的C—C/C=C键;在286.28 eV处的特征峰为S‑PDI的C≡N键;在287.87、288.53、288.75 eV(依次为上、下、中图)处的峰分别归属于NVs、S‑PDI、30% NVs/S‑PDI的 N—C=N。 NVs、S‑PDI、30% NVs/S‑PDI的 N1s谱图如图 3d 所示,398.42 eV(或398.87 eV)处的峰分别归属于NVs(或30% NVs/S‑PDI)的 C—N=C[16];各自谱图中399.98、399.77、400.14 eV处的峰分别归属于NVs、S‑PDI、30% NVs/S‑PDI的N—(C)3键,401.00和400.95 eV处的峰则归属于NVs的C—N—H键,这是由π键激发引起的电荷效应导致的[17]。S‑PDI、30% NVs/S‑PDI的S2p谱图如图3e所示,163.88 eV(或 163.85 eV)属于 S‑PDI(或30% NVs/S‑PDI)的S=C键,结合能的减小可能是由于S‑PDI中的S=C键与NVs之间的相互作用[18]。S‑PDI、30% NVs/S‑PDI的O1s谱图如图 3f所示,531.57 eV(或 531.60 eV)处的峰属于 S‑PDI(或 30% NVs/S‑PDI)的C=O键,531.90 eV(或532.10 eV)处的峰归属于S‑PDI(或30% NVs/S‑PDI)的C—O键,在533.53(或533.49 eV)处的峰归属于S‑PDI(或30% NVs/S‑PDI)的O—H键[19]。由此可见,对应的结合能均发生位移,这是由于在NVs/S‑PDI界面处存在电荷分布差异[20]。
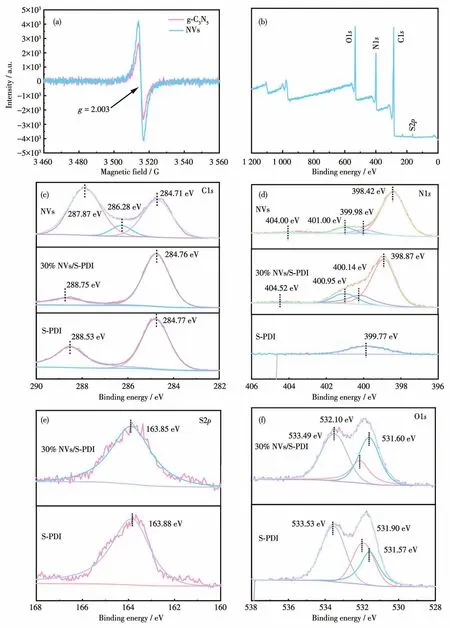
图3 (a)g‑C3N5和NVs的EPR谱图;(b)30% NVs/S‑PDI的XPS全谱图;(c~f)NVs、S‑PDI和30% NVs/S‑PDI的C1s、N1s、S2p、O1s高分辨XPS谱图Fig.3 (a)EPR spectra of g‑C3N5and NVs;(b)Survey XPS spectra of 30% NVs/S‑PDI;(c‑f)High‑resolution XPS spectra of C1s,N1s,S2p,and O1s of NVs,S‑PDI,and 30% NVs/S‑PDI
图4a~4d 是 NVs、S‑PDI和 30% NVs/S‑PDI的TEM图。由图4a、4b可见,NVs由多孔网状结构单元组成,S‑PDI是由纳米片组成;由30% NVs/S‑PDI的TEM图(图4c、4d)可以清晰看到NVs与S‑PDI接触紧密。从能谱面扫(EDS mapping)图可以看出(图4e~4j),30% NVs/S‑PDI存在C、N、O、S四种元素。

图4 (a)NVs和(b)S‑PDI的TEM图;(c、d)30% NVs/S‑PDI的TEM图;(e~j)30% NVs/S‑PDI的EDS mapping图Fig.4 TEM images of(a)NVs and(b)S‑PDI;(c,d)TEM images of 30% NVs/S‑PDI;(e‑j)EDS mappings of 30% NVs/S‑PDI
图5a为所制备样品的UV‑Vis DRS谱图。NVs和S‑PDI的吸收边带分别为504、754 nm。10% NVs/S‑PDI、20% NVs/S‑PDI、30% NVs/S‑PDI、50% NVs/S‑PDI的吸收边带为668、681、698、712 nm,介于NVs与S‑PDI之间。图5b为NVs和S‑PDI的(αhν)1/2‑hν曲线[21],由图可得两者的带隙宽度(Eg)为2.39和1.63 eV。
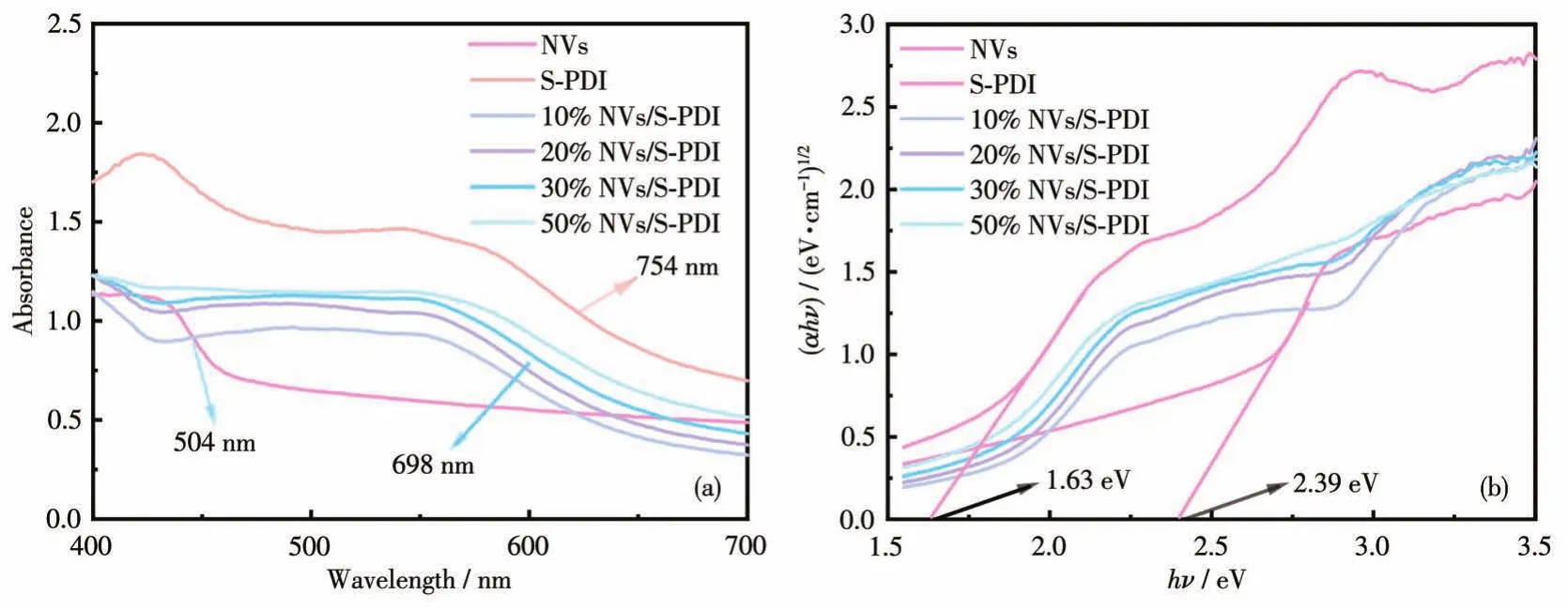
图5 (a)所制备样品的UV‑Vis DRS谱图;(b)NVs和S‑PDI的(αhν)1/2‑hν图Fig.5 (a)UV‑Vis DRS spectra of as‑prepared samples;(b)(αhν)1/2‑hν plots of NVs and S‑PDI
2.2 样品的光催化性能
NVs、S‑PDI和x% NVs/S‑PDI光催化还原 Cr、降解苯酚的性能如图6所示。光照3 h,NVs和S‑PDI对Cr还原率分别为24.88% 和39.33% (图6a);而30% NVs/S‑PDI对Cr还原率可达77.96%,分别是NVs、S‑PDI的3.21倍、2.03倍(图6b)。NVs、S‑PDI光催化还原Cr的表观反应速率常数(kapp)分别为0.095和0.166 h-1(图6c);而30% NVs/S‑PDI光催化还原 Cr的kapp为 0.535 h-1,分别是 NVs、S‑PDI的 5.63倍、3.22倍。光照3 h,NVs和S‑PDI对苯酚的降解率分别为 63.77%和 65.94% (图6d);而 30% NVs/S‑PDI对苯酚的降解率为74.39%,分别是NVs、S‑PDI的1.17倍、1.13倍(图6e)。NVs、S‑PDI光催化降解苯酚的kapp分别为0.338和0.359 h-1(图6f);而30% NVs/S‑PDI的kapp为0.486 h-1,分别是 NVs、S‑PDI的1.44倍、1.35倍。

图6 所制备样品的光催化性能:(a、b)还原Cr;(c)还原Cr的kapp;(d、e)降解苯酚;(f)降解苯酚的kappFig.6 Photocatalytic performance of as‑prepared samples:(a,b)reduction of Cr;(c)kappof reduction of Cr;(d,e)degradation of phenol;(f)kappof degradation of phenol
以苯酚和重铬酸钾混合溶液作为污染物,研究了 NVs、S‑PDI、x% NVs/S‑PDI光催化氧化苯酚耦合Cr还原性能,结果如图7所示。从图7a可知,NVs和S‑PDI对苯酚协同降解率分别为26.32% 、88.66% ;而30% NVs/S‑PDI对苯酚协同降解率达到了93.89%。从图7b可知,NVs和S‑PDI对苯酚协同降解的kapp分别为0.102和0.726 h-1,30% NVs/S‑PDI的kapp则为0.932 h-1,分别是NVs、S‑PDI的9.14倍、1.28倍。从图7c可知,NVs和S‑PDI对Cr协同还原率分别为21.17% 和59.08% ;而30% NVs/S‑PDI对Cr协同还原率为 92.83%,分别是 NVs、S‑PDI的 9.14倍、1.28倍。NVs和S‑PDI对Cr协同还原的kapp分别为0.079和0.297 h-1,30% NVs/S‑PDI对应的kapp为0.878 h-1,分别是NVs、S‑PDI的11.11倍和2.96倍(图7d)。和单独利用催化剂氧化降解苯酚与还原Cr相比,本系统实现了光催化氧化降解苯酚与光催化还原Cr的同步强化。也就是说,苯酚的氧化降解促进了Cr的还原,Cr的还原增强了苯酚的氧化降解。可见,NVs/S‑PDI充分利用了导带的还原性能和价带的氧化性能,实现电子空穴的空间分离,协同强化光催化过程中的氧化半反应和还原半反应,同步提升光催化氧化还原性能。随着光照时间的延长,苯酚和重铬酸钾混合溶液中,苯酚和Cr的浓度逐渐降低(图S2a、S2b)。

图7 在苯酚和重铬酸钾混合溶液体系中,所制备样品的光催化性能:(a)降解苯酚;(b)降解苯酚的kapp;(c)还原Cr;(d)还原Cr的kappFig.7 Photocatalytic performance of as‑prepared samples in the mixed solution of phenol and potassium dichromate:(a)degradation of phenol;(b)kappof degradation of phenol;(c)reduction of Cr;(d)kappof reduction of Cr
此外,研究了NVs、S‑PDI、x% NVs/S‑PDI产H2O2的性能,结果如图8所示。由图8a可知,不加任何牺牲剂时,可见光照射2 h后NVs不能产出H2O2,S‑PDI的 H2O2产量为 15.88 μmol·L-1,20% NVs/S‑PDI的H2O2产量为 30.68 μmol·L-1,是 S‑PDI的 1.94倍。由图 8b 可知,NVs对 H2O2的产出速率为 0 μmol·L-1·h-1,S‑PDI的为 7.94 μmol·L-1·h-1,而 20% NVs/S‑PDI的为 15.34 μmol·L-1·h-1,是 S‑PDI的1.93倍。由图8c可知,改变溶液的pH,对20% NVs/S‑PDI产出H2O2的影响不大。
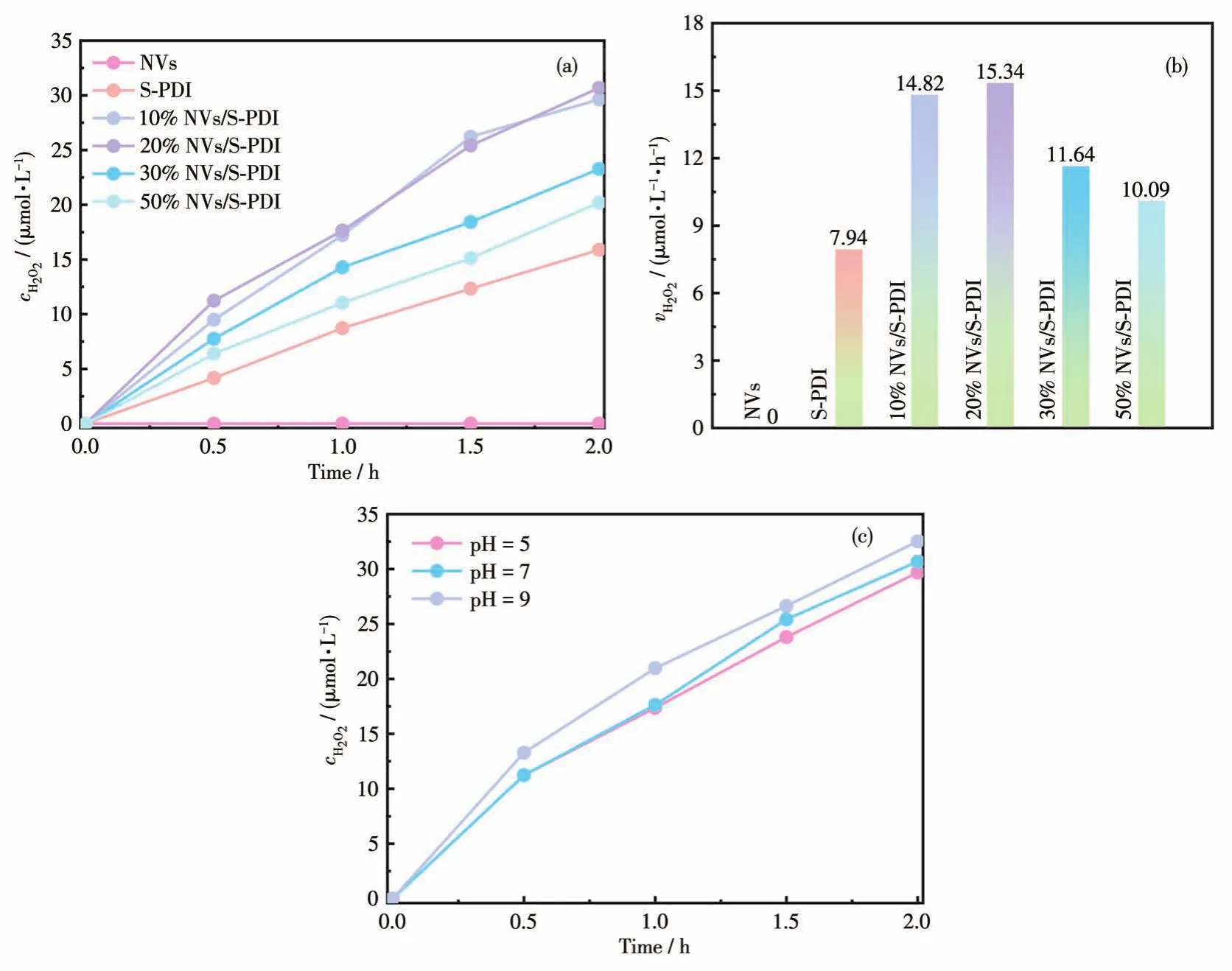
图8 所制备样品的(a)H2O2产量和(b)vH2O2;(c)20% NVs/S‑PDI在不同pH值时的H2O2产量Fig.8 (a)Yield of H2O2and(b)vH2O2over as‑prepared samples;(c)Yield of H2O2over 20% NVs/S‑PDI at different pH values
分别采用BQ、IPA、EDTA‑2Na捕获30% NVs/S‑PDI协同氧化苯酚与还原Cr过程中产生的、·OH和h+,结果如图9a所示。加入BQ时,30% NVs/S‑PDI协同氧化苯酚性能明显降低,降解率为18.91%。加入IPA时,30% NVs/S‑PDI协同氧化苯酚性能变化不大,降解率为77.14%。加入EDTA‑2Na时,30% NVs/S‑PDI协同氧化苯酚效果明显降低,降解率为33.05%。因此,氧化降解苯酚的活性物种主要是·O2-和h+。D MPO‑·O2-的ESR谱图如图9b所示。由图可知,黑暗中并未检测到·O2-,光照4 min,30% NVs/S‑PDI出现了DMPO‑·O2-特征信号,而光照8 min,特征信号峰加强。DMPO‑h+的ESR谱图如图9c所示。黑暗条件下出现了DMPO‑h+信号,随着光照时间的延长,特征信号峰加强。因此,可见光照射30% NVs/S‑PDI时产生·O2-和 h+。

图9 30% NVs/S‑PDI协同氧化苯酚与还原Cr机理的探究:(a)活性自由基捕获;(b)DMPO‑·O2-和(c)DMPO‑h+的ESR谱图;分别加入(d)IPA、(e)BQ、(f)EDTA‑2Na后的HPLC图Fig.9 Investigation on mechanism of synergistic oxidation of phenol and reduction of Cr by 30% NVs/S‑PDI:(a)trapping of active radical;ESR spectra of(b)DMPO‑·O2‑and(c)DMPO‑h+;HPLC diagrams upon the addition of(d)IPA,(e)BQ,and(f)EDTA‑2Na,respectively
在苯酚和重铬酸钾混合溶液中分别加入BQ、IPA和EDTA‑2Na捕获剂,探索了30% NVs/S‑PDI协同氧化苯酚与还原Cr过程中苯酚的降解中间产物,结果如图9所示。当加入IPA时(图9d),在tD为1.98、3.98 min处出现特征峰,分别对应羧酸、BQ,且随着反应的进行,羧酸、BQ的峰逐渐减小。可见,在·O2-和空穴存在下,苯酚被氧化成BQ,进一步氧化为羧酸,最后被矿化为 CO2、H2O(图 S3,步骤 1)。当加入BQ时(图9e),在tD为3.98、3.23和1.98 min处出现特征峰,分别对应BQ、对苯二酚、羧酸,初始时BQ的浓度为0.1 mmol·L-1,且随着反应时间的延长,部分苯酚被氧化为BQ,BQ的浓度先增加后下降(图S4),BQ进一步转化为对苯二酚,则对应对苯二酚的峰逐渐增大。可见,在·OH和空穴存在下,苯酚首先被氧化成BQ,随着反应时间的进行,进一步氧化成对苯二酚,之后氧化为羧酸,最后被矿化成CO2、H2O(图S3,步骤2)。当加入EDTA‑2Na时(图9f),在tD为1.98、2.40 min处出现特征峰,分别对应羧酸、丙醇,且随着反应的进行,羧酸和丙醇的峰逐渐减小[22]。因此,在·O2-和·OH的存在下,苯酚氧化成对苯二酚,然后开环形成丙醇,进一步被氧化为羧酸,之后被矿化成 CO2、H2O(图S3,步骤3)。此外,在苯酚的降解路径中,苯酚的降解产物羧酸或丙醇被催化剂的价带空穴氧化,而与价带空穴成对的导带电子则将Cr还原为Cr。因此,在30% NVs/S‑PDI协同氧化苯酚与还原Cr过程中,苯酚降解为羧酸、丙醇、对苯二酚和BQ,而中间产物羧酸、丙醇可以增强Cr的还原,苯酚的氧化与Cr的还原反应得到互相促进。
30% NVs/S‑PDI的电化学阻抗谱图和光致发光(PL)谱图如图10所示。NVs的阻抗圆弧最大,而30% NVs/S‑PDI的阻抗圆弧最小(图10a)。30% NVs/S‑PDI的 PL强度最小(图 10b),表明 30% NVs/S‑PDI具有高的电子-空穴的分离效率。以饱和甘汞电极作为参比电极测量了 NVs和 S‑PDI的 Mott‑Schottky曲线,结果如图10c、10d所示。NVs和S‑PDI的平带电位分别为-1.60和-1.06 V。NVs和S‑PDI的Mott‑Schottky曲线的斜率均为正值,表明二者均为n型半导体。因此,NVs和S‑PDI的相对于标准氢电极(NHE)的平带电位分别为-1.36和0.82 V。对于n型半导体,导带电位与平带电位的差值为0.1 V[23]。所以,NVs和S‑PDI的导带分别为-1.46和-0.92 eV。
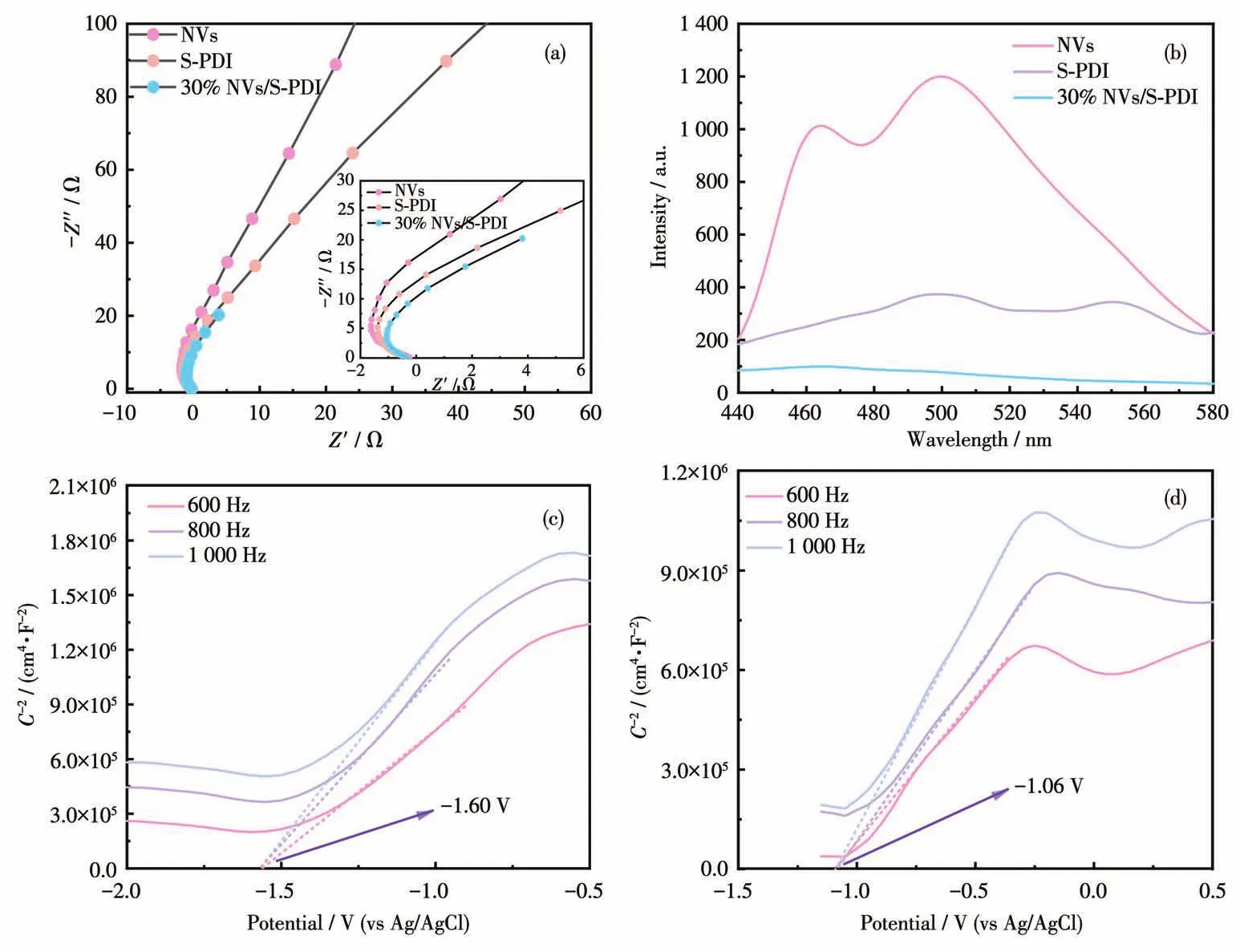
图10 NVs、S‑PDI和30% NVs/S‑PDI的(a)电化学阻抗谱图和(b)PL谱图;(c)NVs和(d)S‑PDI的Mott‑Schottky曲线Fig.10 (a)Electrochemical impedance spectra and(b)PL spectra of NVs,S‑PDI,and 30% NVs/S‑PDI;Mott‑Schottky curves of(c)NVs and(d)S‑PDI
由于NVs的导带位置低于S‑PDI的导带位置,而NVs的价带位置高于S‑PDI的价带位置,因此,30% NVs/S‑PDI协同氧化苯酚与还原Cr的可能机理如图11所示。在光照条件下,NVs和S‑PDI产生电子、空穴(反应方程式1、2)。空穴由NVs的价带迁移至S‑PDI的价带,同时一部分Cr在NVs的导带上得到电子还原为Cr。在S‑PDI的价带上,空穴将苯酚氧化成丙醇、羧酸,进而被矿化形成CO2、H2O等(反应方程式3)。电子由NVs的导带迁移至S‑PDI的导带。同时S‑PDI的导带低于O2/·O2-(-0.33 eV),表明O2可以在S‑PDI的导带上还原为·O2-(反应方程式 4);在 S‑PDI导带,Cr得到电子还原为 Cr,同时H2O还原为·OH(反应方程式5)。由于S‑PDI的导带(-0.92 eV)低于 O2/H2O2(0.68 eV),可以将 O2转化成H2O2(反应方程式6)。一方面,丙醇和羧酸可以将Cr还原为Cr(反应方程式7);另一方面,光照产生的电子、Cr、H2O2形成一个光自芬顿反应过程[24](反应方程式8),可以进一步将苯酚氧化降解(反应方程式9)。
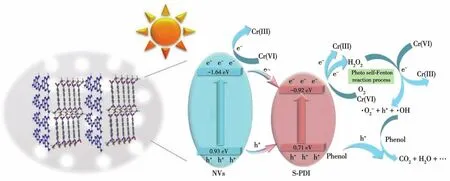
图11 30% NVs/S‑PDI协同氧化苯酚与还原Cr的机理Fig.11 Mechanism of synergistic oxidation of phenol and reduction of Cr by 30% NVs/S‑PDI

3 结论
通过静电自组装制备有机复合半导体N缺陷g‑C3N5修饰S掺杂PDI(NVs/S‑PDI)。N缺陷使g‑C3N5具有丰富的活性位点,而具有氨基基团的酰胺增强了S‑PDI与NVs的分子间作用力。30% NVs/S‑PDI协同氧化苯酚与还原Cr过程中,Cr的还原率为92.83%,分别是S‑PDI、NVs的2.95倍、11.11倍;苯酚的降解率为93.89%,分别是S‑PDI、NVs的1.28倍、8.60倍;同时也是30% NVs/S‑PDI单独氧化苯酚时降解率的1.26倍,是30% NVs/S‑PDI单独还原Cr时还原率的1.19倍。NVs/S‑PDI充分利用了两者的导带还原性能和价带氧化性能,使电子空穴空间分离得以实现,协同强化光催化过程中的氧化半反应和还原半反应,同步提升苯酚的降解和Cr的还原。此外,光照产生的电子、H2O2与Cr形成一个光自芬顿反应,促进了苯酚的降解与Cr的还原。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
吉首大学学报(自然科学版)(2023年6期)2023-12-22 08:18:20
云南化工(2021年10期)2021-12-21 07:33:28
装备维修技术(2021年36期)2021-10-25 13:21:04
弹箭与制导学报(2021年3期)2021-07-30 02:56:52
建材发展导向(2021年24期)2021-02-12 02:00:02
网印工业(2019年4期)2019-05-21 06:41:58
重型机械(2019年2期)2019-04-28 11:52:04
吉首大学学报(自然科学版)(2018年3期)2018-07-03 03:14:12
Chinese Journal of Chemical Engineering(2017年5期)2017-05-28 10:22:54
中国塑料(2014年1期)2014-10-17 02:46:34
