
冠状动脉粥样硬化性心脏病患者心外膜脂肪组织的生物信息学研究
2023-02-01 09:04柴晏赵玉青郭旭男王东英边云飞
中国全科医学 2023年8期
柴晏,赵玉青,郭旭男,王东英,边云飞
动脉粥样硬化(atherosclerosis,AS)导致的心血管疾病(cardiovascular disease,CVD)是全球死亡的主要原因,世界卫生组织预测2030年将有2 360万人死于CVD[1]。AS形成机制复杂,主要涉及血管内皮损伤、单核巨噬细胞黏附、脂质沉积等[2],但这些因素如何影响AS进程,其机制尚未完全清楚。因此找寻AS新的诊断靶点,预防AS的发生,延缓其进展尤为重要。利用基因芯片技术对临床患者标本进行检测分析,筛选出一些具有价值的基因,进而深入研究冠状动脉粥样硬化性心脏病(coronary artery disease,CAD)的发病机制具有重要的临床意义。肥胖是CVD的独立危险因素,但肥胖的主要评价指标体质指数(BMI)与AS患者死亡率呈“U”型关联,这种现象被称为肥胖悖论,因此脂肪组织不再被认为是单纯的“能量仓库”,而是一种代谢活跃的内分泌和旁分泌器官[3]。心外膜脂肪组织(epicardial adipose tissue,EAT)在解剖上与冠状动脉紧密相连,同AS、心房颤动、心力衰竭等CVD密切相关[4-6]。多项研究发现功能失调的EAT通过分泌外泌体(exosome,EXO)和生物活性物质促进AS疾病进展[7],但其作用机制仍需进一步研究。本研究通过对美国国家生物技术信息中心(NCBI)的基因表达综合数据库(Gene Expression Omnibus database,GEO)中 CAD患者EAT和EXO的基因测序数据进行生物信息学分析,探讨EAT参与AS的分子机制及其与EXO的联系,旨在为CAD的诊断和治疗提供新的理论基础。
1 材料与方法
1.1 数据集的下载与预处理 以“epicardial adipose tissue OR EAT”为关键词,选择“Homo sapiens”为研究对象检索GEO数据库,根据数据集提供的相关信息,获得所需芯片GSE64554、GSE120774,根据临床信息将EAT的测序数据分为CAD组和健康对照组。下载series matrix的数据,采用“sva”包“ComBat”方法去除批次效应,合并基因表达矩阵。通过exoRBase 2.0数据库获取CAD组与健康对照组血液EXO基因表达谱。
1.2 差异基因的筛选 通过“limma”R语言包[8]对合并后数据集中CAD组与健康对照组EAT间差异表达基因(differentially expressed genes,DEGs)和CAD组与健康对照组EXO间DEGs进行筛选,以P<0.05为筛选条件,并将结果可视化为火山图和热图。
1.3 加权基因共表达网络(WGCNA)的构建与CAD相关模块的识别 为了防止合并数据集对基因关联度的影响,使用“WGCNA”R语言包构建GSE64554数据集基因共表达网络并识别出模块。选择满足无尺度网络的标准软阈值β构建基因共表达网络,转化拓扑重叠矩阵(topological overlap matrix,TOM),采用动态剪枝法对模块进行初步划分。根据GSE64554数据集样本临床信息,决定纳入的临床信息包括疾病和年龄,得到相关性最高的模块进行后续分析。计算基因与模块的相关系数(module membership,MM)来评估基因与模块之间的相关性,以|MM|>0.8筛选模块内枢纽基因(hub gene)。
1.4 基因功能的富集分析 通过“clusterProfiler”R语言包[9]将所获基因进行富集分析,以FDR<0.05为筛选标准,进行通路富集分析。通过GO富集分析,得到所选基因所富集的生物过程(biological process,BP)、细胞组分(cellular components,CC)及分子功能(molecular function,MF)。通过KEGG通路富集分析,得到所选基因所富集的信号通路。将所获基因导入Metascape在线数据库[10]进行基因的富集分析以及转录因子预测。
1.5 蛋白质-蛋白质相互作用(protein-protein interaction,PPI)网络的构建与hub 基因筛选 将所选基因导入STRING数据库(https://string-db.org)构建PPI网络,将PPI网络导入Cytoscape(Version 3.9.0)软件[11]通过CytoHubba插件MCC算法获得连接度最高的10个基因作为关键基因,并进行绘图。
1.6 免疫细胞浸润分析 在Cibersort网站(https://cibersort.stanford.edu/)下载22种免疫细胞的基因表达矩阵,采用Cibersort反卷积算法,计算GSE64554芯片中46个样本的免疫细胞浸润情况,并作图对组织的免疫细胞浸润情况进行比较。
1.7 临床样本的采集及实时荧光定量PCR(quantitative real-time PCR,qRT-PCR) 选取山西医科大学第二医院2021年6—10月行冠状动脉造影患者20例,其中CAD患者及健康者各10例,留取外周血储存在-80 ℃冰箱中备用。引物由武汉赛维尔生物科技有限公司合成,见表1。按照总RNA快速提取试剂盒(RP4001,百泰克公司)的说明提取血浆中的总RNA,cDNA的合成按照HiScriptⅢ RT SuperMix for qPCR(+gDNA wiper)试剂盒的步骤进行,第一步去除总RNA中残余的DNA,第二步反转录cDNA,得到的cDNA保存于-20 ℃冰箱。以cDNA为模板,进行qRT-PCR,反应程序为:95 ℃3 min;95 ℃ 5 s;60 ℃ 10 s,72 ℃ 30 s,40 个循环;95 ℃15 s,65 ℃ 5 s,95 ℃ 50 s。结果用2-ΔΔCT进行计算分析。
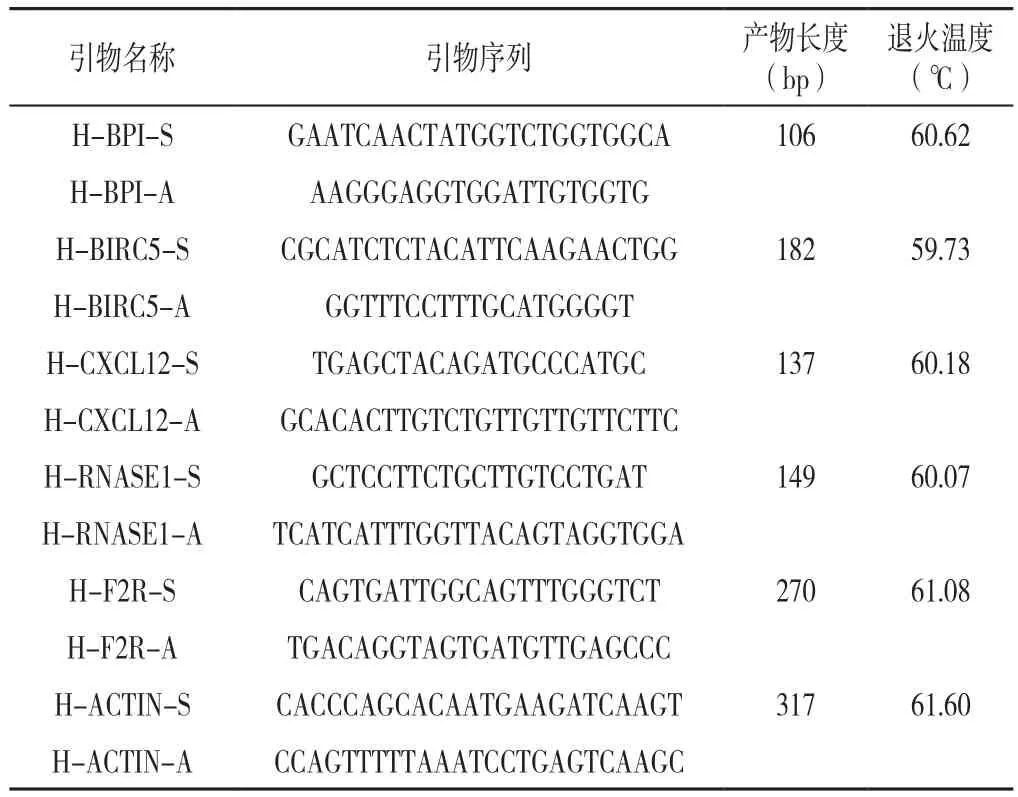
表1 PCR引物Table 1 Primer sequences for qRT-PCR
1.8 统计学方法 采用Excel对录入整理数据,应用SPSS 26.0软件对数据进行统计学分析,符合正态分布的连续变量采用(±s)表示,两组间比较采用独立样本t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 CAD组与健康对照组EAT间的DEGs
2.1.1 DEGs的获取 采用“limma”包获取CAD组与健康对照组EAT间DEGs,以P<0.05作为筛选差异基因的阈值,得到差异表达的1 511个mRNA,其中表达上调956个,表达下调555个,见图1、2。

图1 CAD组与健康对照组EAT间DEGs火山图Figure 1 Volcano plot of differentially expressed genes in epicardial adipose tissue between CAD group and healthy control group

图2 CAD组与健康对照组EAT间DEGs热图Figure 2 Heatmap of differentially expressed genes in epicardial adipose tissue between CAD group and healthy control group
2.1.2 EAT间DEGs的GO/KEGG富集分析与PPI网络对合并后CAD组较健康对照组EAT间DEGs进行GO富集分析,共获得符合筛选条件的GO术语共800条,其中BP 567条,主要涉及细胞压力反应、细胞DNA损伤刺激反应等。获得CC 127条,主要涉及细胞质基质、MHC蛋白复合物、内质网腔侧的组成部分等。获得MF 106条,主要涉及MHCⅡ类蛋白复合物、特殊核糖核酸酶激活等,见图3。共富集到KEGG通路20个,主要涉及RNA降解、病毒性心肌炎、移植物抗宿主病、1型糖尿病等信号通路,见图4。
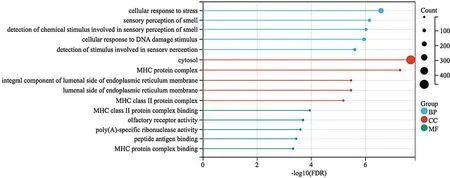
图3 EAT间DEGs的GO富集分析Figure 3 GO enrichment analysis of differentially expressed genes in epicardial adipose tissue
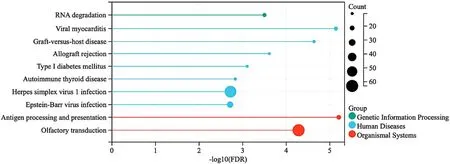
图4 EAT间DEGs的KEGG富集分析Figure 4 KEGG enrichment analysis of differentially expressed genes in epicardial adipose tissue
将所获EAT间DEGs输入STRING数据库,构建PPI网络,其中实际上有互作关系的节点有1 507个,有1 258条边,每个节点的平均Degree为1.67分。将PPI网络图导入Cytoscape软件,使用CytoHubba插件根据MCC算法得到连接度前10的基因并作图,分别为 RPS27A、PSME4、PSMA1、PSMA5、GLI3、GLI1、PSME2、PSMB9、SUFU和SHH,见图5。

图5 EAT间DEGs的PPI网络与关键基因Figure 5 Differentially expressed genes in epicardial adipose tissue and the key genes identified in the PPI network
2.1.3 EAT间DEGs的Metascape富集分析 对合并后CAD组与健康对照组EAT间DEGs进行Metascape富集分析,发现DEGs主要富集于嗅觉转导、细胞对DNA损伤刺激反应、单纯疱疹病毒1型感染、RNA代谢、调节细胞对压力的反应、适应性免疫系统等,TRRUST数据库预测MHCⅡ反式激活蛋白(CIITA)转录因子可能参与了EAT间DEGs的调控,见图6、7。

图6 EAT间DEGs的Metascape富集分析Figure 6 Metascape enrichment analysis of differentially expressed genes in epicardial adipose tissue
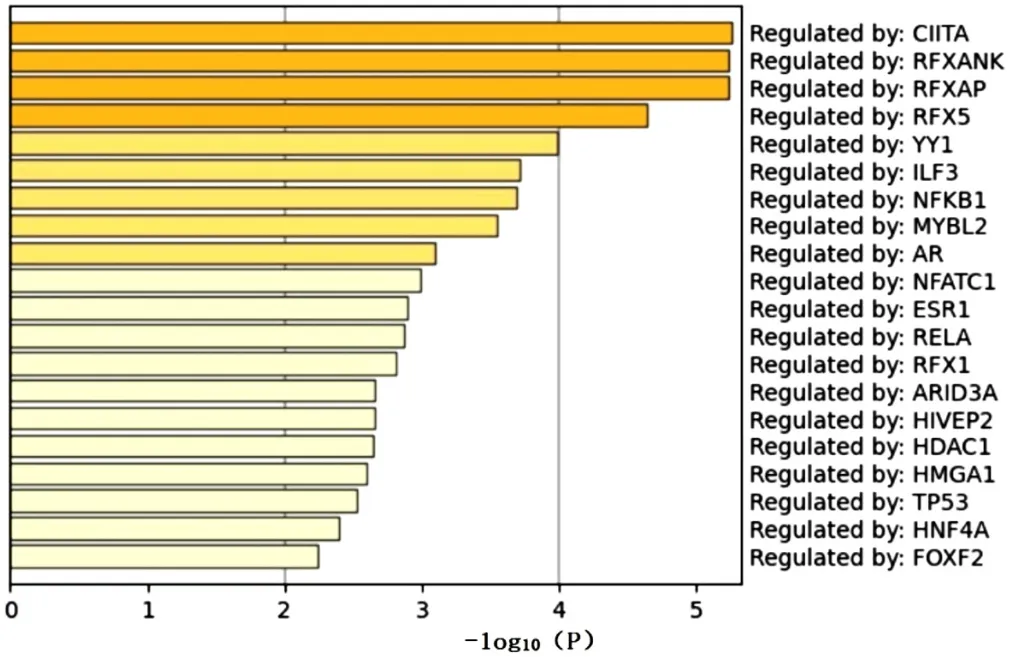
图7 EAT间DEGs的TRRUST数据库预测转录因子Figure 7 TRRUST database-based analysis of differentially expressed genes in epicardial adipose tissue to predict transcription factors
2.2 EAT组织的WGCNA分析
2.2.1 EAT组织WGCNA软阈值的选择与模块的初步划分 本研究最终以β=5作为软阈值来构建共表达网络,设置每个基因模块中基因数目最小为30,设定基因的聚类高度上限为0.995。最终得到156个网络模块,模块中的基因个数为30~2 169。根据拓扑重叠程度,制作模块相关性图,见图8、9。

图8 软阈值参数网络拓扑结构的分析Figure 8 Network topology for soft thresholding powers
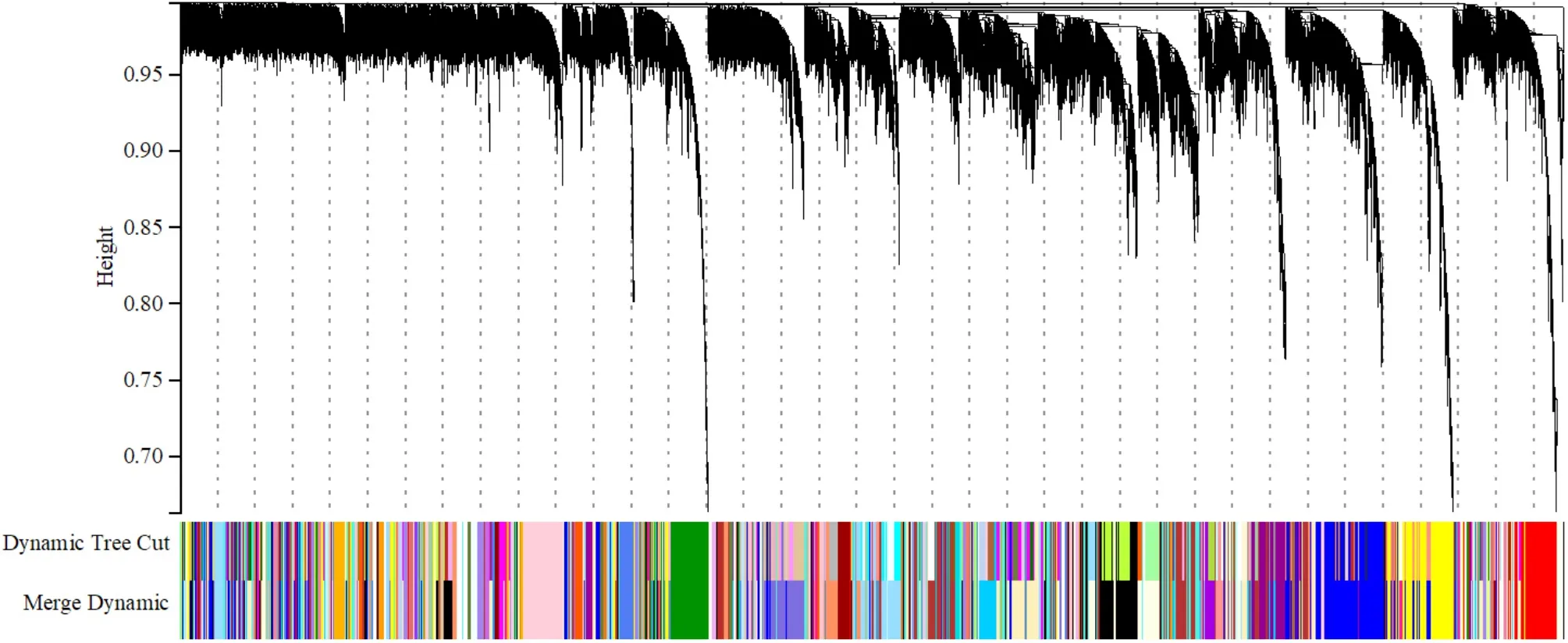
图9 基因聚类树状图Figure 9 Dendrogram of gene clustering
2.2.2 临床信息关联与hub基因的获取 将各个划分出的模块与临床信息进行关联分析,得到与AS患者的EAT样本临床信息最为相关的模块进行后续研究。结果发现,AS患者EAT与green4(r=0.50,P=0.02)、slateblue(r=0.54,P=8.0×10-3)、darkseagreen4(r=0.55,P=6.4×10-3)、darkolivegreen1(r=0.55,P=7.0×10-3)、sienna4(r=0.56,P=5.6×10-3)、brown3(r=0.57,P=4.2×10-3)模块呈正相关,与 deeppink(r=0.50,P=0.01)、lavender(r=0.60,P=2.6×10-3)、lavenderblush3(r=0.56,P=5.9×10-3)呈负相关,以|MM|>0.8筛选所选模块内hub基因,见图10。
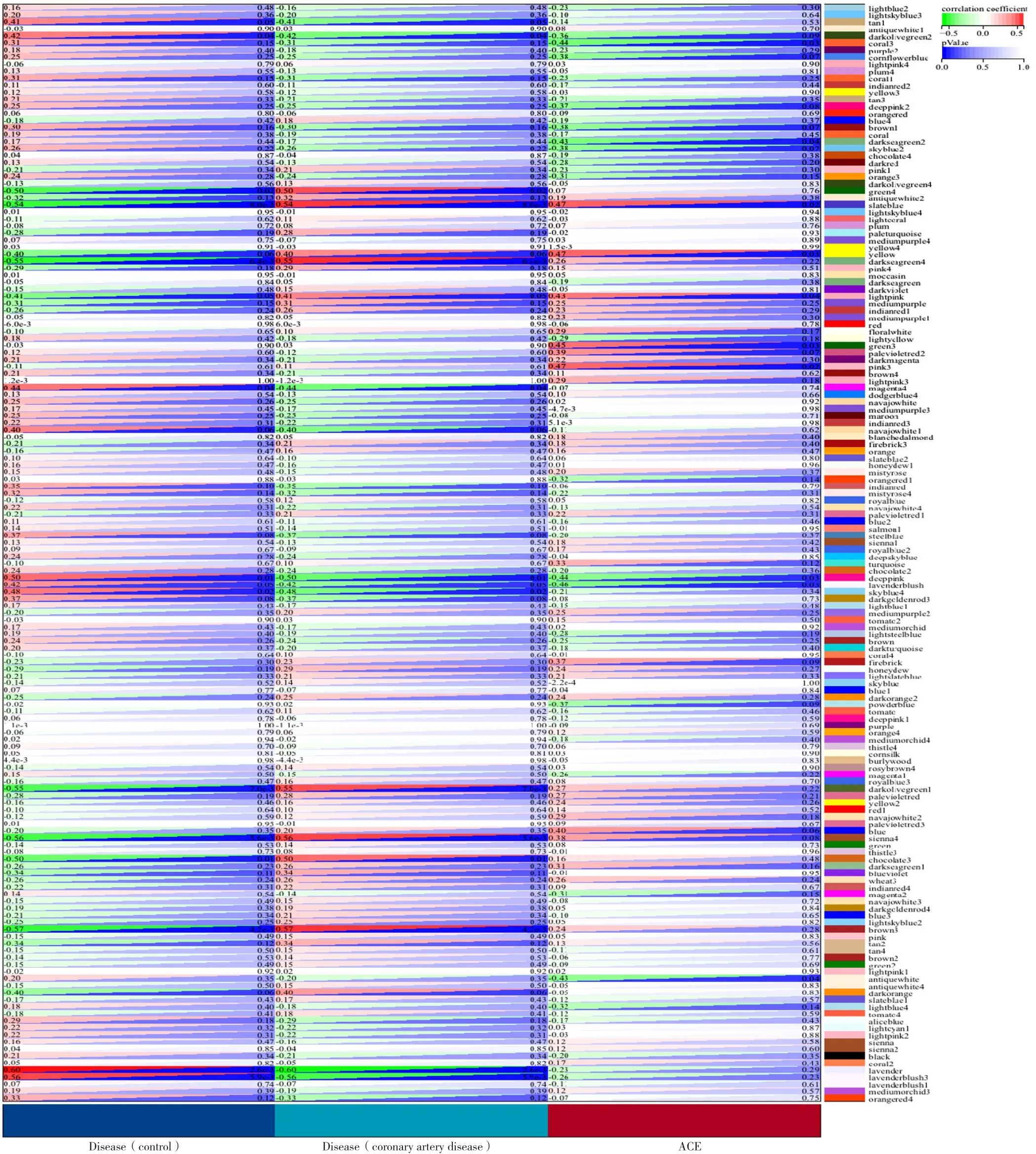
图10 基因模块与临床信息关联性Figure 10 Correlation between gene modules and clinical information
2.3 关键基因的获得 将DEGs同模块内hub基因取交集,获得差异表达的模块内hub基因,将其作为EAT组织参与AS病理生理过程的关键基因,其中上调hub基因为DDX47、FEM1C、NOL11、SRP54、ABI1、PATL1、BNIP2,下调hub基因为C1orf159、CHCHD4,见图11。
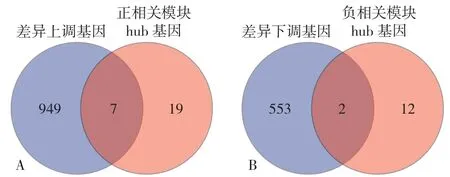
图11 差异性表达基因与模块内hub基因Venn图Figure 11 Venn diagram of differentially expressed genes and hub genes in the module
2.4 免疫细胞浸润分析 通过Cibersort反卷积算法,计算GSE64554芯片中CAD组和健康对照组EAT的免疫细胞浸润情况。研究发现,CAD组EAT组织较健康对照组中初始CD4+T细胞表达丰度升高(P=0.048),静息树突细胞表达丰度减低(P=0.044),见图12、13。

图12 GSE64554数据集EAT组织免疫细胞浸润柱状图Figure 12 Bar graph of immune cell infiltration in epicardial adipose tissue between CAD patients and controls included in dataset GSE64554

图13 GSE64554数据集EAT组织免疫细胞浸润小提琴图Figure 13 Violin plot of immune cell infiltration in epicardial adipose tissue between CAD patients and controls included in dataset GSE64554
2.5 CAD组与健康对照组EXO间DEGs 采用“limma”包选获取CAD组与健康对照组EXO间DEGs,以P<0.05,|logFoldchange|>1作为筛选差异基因的阈值,共得到DEGs 1 658个,其中上调278个,下调1 380个,见图14、15。

图14 CAD组与健康对照组EXO间火山图Figure 14 Volcano plot of differentially expressed genes in exosomes between CAD patients and heathy controls
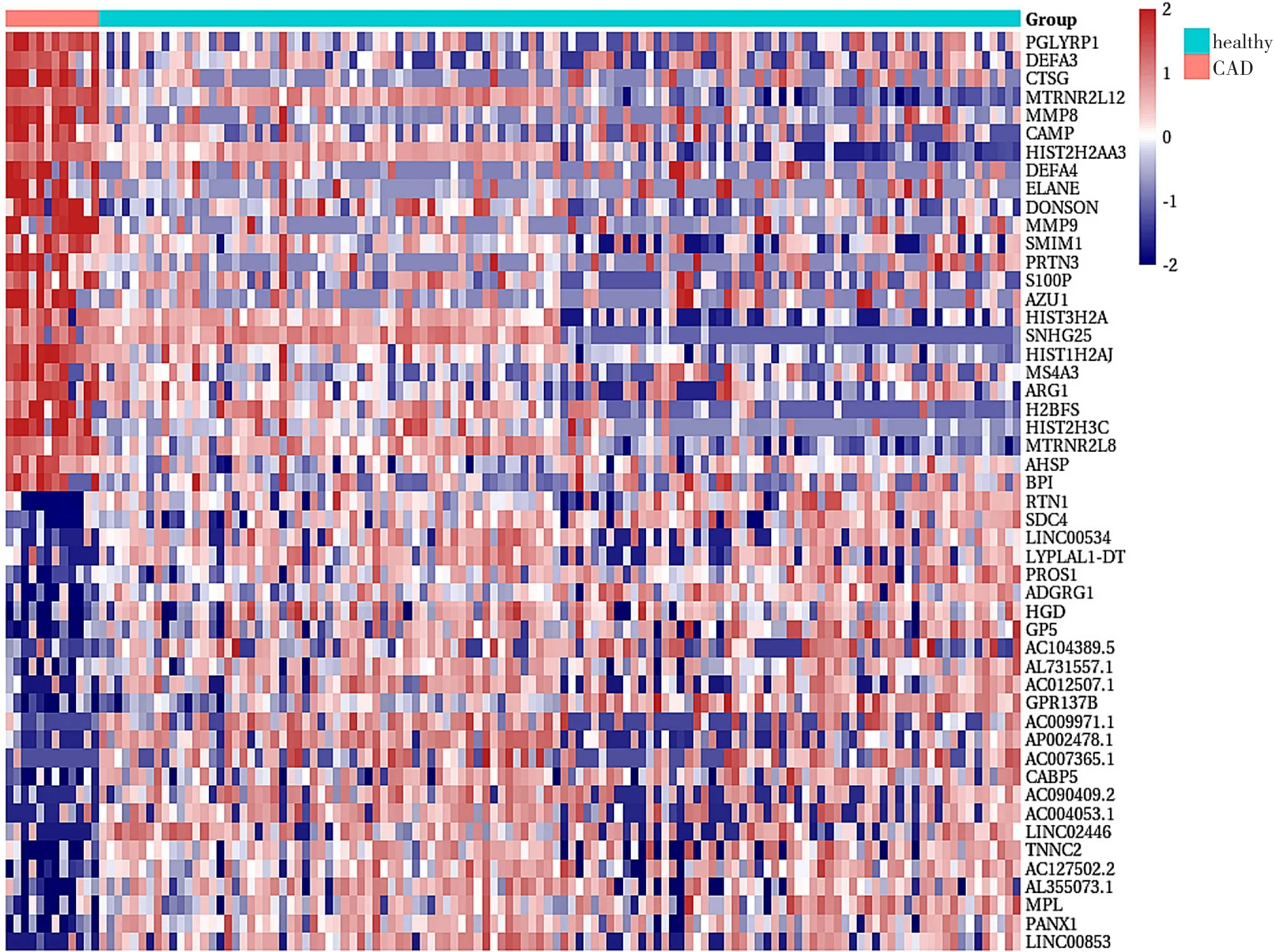
图15 CAD组与健康对照组EXO间DEGs热图Figure 15 Heatmap of differentially expressed genes in exosomes between CAD patients and heathy controls
2.6 EAT来源EXO 将CAD组与健康对照组EAT、EXO间DEGs取交集并做Venn图,共获得129个基因,其中上调16个,下调113个,经筛选,将BPI、BIRC5、CXCL12、RNASE1、F2R作为关键基因并通过qRT-PCR进行验证,见图16。
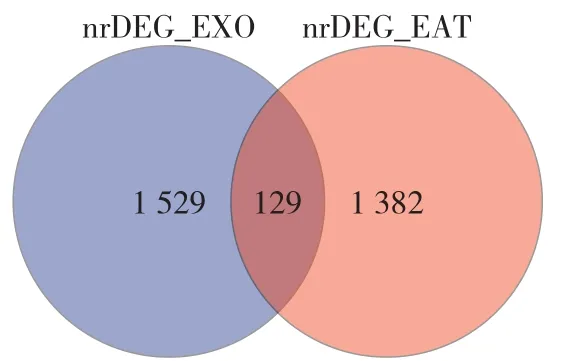
图16 CAD患者EAT组织、EXO间DEGs Venn图Figure 16 Venn diagram of differentially expressed genes in epicardial adipose tissue and exosomes from CAD patients
2.7 EAT来源EXO间DEGs的验证 收集CAD组和健康对照组外周血,运用qRT-PCR验证生物信息学分析所得的EAT来源EXO基因BPI、BIRC5、CXCL12、RNASE1、F2R的mRNA水平。结果显示,与健康对照组相比,CAD患者的5个所选基因的表达水平差异有统计学意义(P<0.05),其中BPI、BIRC5、CXCL12和RNASE1的mRNA水平升高,F2R基因的mRNA水平下降(P<0.05),见表2。
表2 CAD组与健康对照组EXO差异性基因表达(±s)Table 2 Differentially expressed genes in exosomes between CAD patients and healthy controls

表2 CAD组与健康对照组EXO差异性基因表达(±s)Table 2 Differentially expressed genes in exosomes between CAD patients and healthy controls
注:CAD=冠状动脉粥样硬化性心脏病;BPI、BIRC5、CXCL12、RNASE1、F2R为心外膜、EXO间DEGs
组别 样本数 BPI BIRC5 CXCL12 RNASE1 F2R CAD 组 10 2.14±0.32 1.98±0.23 2.91±0.89 4.47±1.56 0.45±0.06健康对照组 10 0.99±0.17 1.02±0.11 1.14±0.44 1.28±0.56 1.00±0.12 t值 -9.945 -12.070 -5.630 -6.001 12.637 P值 0.011 0.010 <0.001 0.009 <0.001
3 讨论
EAT作为一种特殊的脂肪组织,解剖结构上紧贴冠状动脉,在病理环境中其向炎症表型转化,通过分泌促炎细胞因子和EXO参与血管内皮损伤、血管重塑、免疫细胞黏附等AS病理生理过程。EAT的炎性反应是AS的全新诊断治疗靶点[5],OIKONOMOU等[12]通过影像组学技术将心外膜脂肪衰减系数(fat attenuation index,FAI)应用于CAD患者的早期筛查,而目前已广泛应用于临床的钠-葡萄糖协同转运蛋白2(sodiumdependent glucose transporters 2,SGLT-2)抑制剂达格列净则可以通过减轻EAT炎症而减少糖尿病患者心血管事件的发生率[13],但EAT在CAD中的作用机制仍需进一步研究。
此次通过生物信息学分析的方法从数据集GSE64554和GSE120774中筛选出CAD组和健康对照组EAT间DEGs 1 511个,通过富集分析发现DEGs主要富集于细胞质基质、MHC蛋白复合物、MHCⅡ蛋白复合物、RNA降解、抗原加工和呈递等。目前AS被认为是一种慢性炎症性疾病,MCHⅡ作为T细胞的呈递抗原,通过抗原识别、细胞活化、产生细胞因子等方式在炎症过程和CAD进展中发挥作用[14]。通过Meatscape富集分析中的TRRUST数据库,笔者预测到CIITA转录因子可能在EAT中发挥了重要的调节作用。研究发现,CIITA通过其转录后修饰甲基化[15]、去乙酰化[16-17]调节MHCⅡ的转录而参与AS。通过免疫细胞浸润分析本研究发现CAD患者EAT中幼稚CD4+T细胞丰度升高,早在2015年IBORRA等[18]便指出可以通过观察幼稚CD4+T细胞的体外分化程度评估AS程度,GADDIS等[19]指出AS通过影响糖酵解而损害幼稚CD4+T细胞功能。综上所述,MHCⅡ在AS中发挥了重要作用,但其在CAD患者EAT中的作用和调控机制仍需进一步研究。
通过筛选CAD患者与健康对照组EAT间DEGs,构建EAT组织WGCNA,获得了DDX47、FEM1C、NOL11、SRP54、ABI1、PATL1、BNIP2、C1orf159、CHCHD4、RPS27A共10个基因作为EAT参与CAD病理生理过程的关键基因。目前仅有关键基因被发现在CVD中发挥作用,XIA等[20]通过生物信息学技术发现免疫相关基因RPS27A可能参与AS的病理过程,HUANG等[21]指出RPS27A基因在糖尿病视网膜病变的关键基因且同幼稚CD4+T细胞密切相关。LI等[22]发现MBNL1通过对ABI1的剪切调控AS中平滑肌细胞的表型转换。因此,关键基因仍需进一步进行细胞和动物实验证明其在EAT中的差异表达以及相关功能。
内皮细胞、平滑肌细胞、心肌细胞、脂肪细胞和血小板来源的EXO通过转运蛋白质、RNA、DNA和其他生物活性物质在缺血再灌注损伤、血管钙化、AS和心脏重塑中发挥了重要作用,已成为CAD的全新诊断治疗靶点[23]。脂肪组织是循环中EXO的来源之一[24],冠状动脉血管周围脂肪组织是一种特殊的EAT[25],LIU等[26]发现血管周围脂肪组织来源EXO中miR-382-5p通过ABCA1/ABCG1信号通路调节泡沫巨噬细胞形成。LI等[27]发现血管周围脂肪组织来源EXO中miR-221-3p通过介导血管重塑参与AS。ZHAO等[7]发现芒果苷诱导血管周围脂肪组织衍生的EXO可以改善AS中的内皮功能障碍,因此EAT来源EXO在AS中的作用具有重要的研究价值。
为进一步探讨EAT在CAD中的作用机制,本研究将CAD患者与健康对照组的EAT间DEGs、EXO间DEGs取交集,共获得129个基因,并筛选表达程度较高、有意义的基因RNASE1、BPI、BIRC5、CXCL12、F2R。其中前4个基因为CAD患者中上调基因。RNASE1是一种可分泌的耐热蛋白,具有强大的心脏保护功能[28],其通过保护内皮细胞在炎症中的损伤而成为血管稳态的关键调控者[29],在CAD中,EAT可能通过EXO释放RNASE1进而调节冠状动脉内皮细胞的炎性损伤。血管生成趋化因子介导细胞趋化、白细胞脱粒和血管生成,CXCL-12同AS密切相关[30],其通过CXCL-12-CXCR4轴参与平滑肌细胞表型转化保护血管稳态[31],特异性过表达巨噬细胞IGF-1通过减少CXCL-12介导的单核细胞募集和增加ABCA1依赖性巨噬细胞脂质外流增加AS斑块的稳定性[32],但目前尚无关于EXO内CXCL-12的相关研究。BIRC5是凋亡抑制蛋白(IAP)家族的成员,LI等[33]通过对小鼠颈动脉单细胞测序发现BIRC5可能与血流紊乱所致血管巨噬细胞聚集相关。早在2002年便有报道指出BPI的单核苷酸多态性同心肌梗死密切相关[34],BLEIJERVELD等[35]通过循环中粒细胞的深度蛋白质组分析发现了BPI可以作为严重AS性冠状动脉狭窄的生物标志物,这表明BPI可以作为晚期CAD乃至预测急性心血管事件的标志物。F2R是血管壁炎症和血栓形成的关键调节剂,其多态性同氯吡格雷疗效密切相关[36],但目前少有F2R在AS中的研究。
综上所述,本研究共确定了DDX47、FEM1C、NOL11、SRP54、ABI1、PATL1、BNIP2、C1orf159、CHCHD4、RPS27A共10个关键基因,RNASE1、BPI、BIRC5、CXCL12、F2R共5个可能源自EAT EXO的基因进行了验证,证明其在CAD中的诊疗意义,此外还探讨了CAD患者EAT免疫浸润情况,为CAD的基础研究提供理论支持。本研究有其局限性,本研究主要针对mRNA,但蛋白质才是生命活动的执行者,mRNA层面的改变并不能等同蛋白质层面的改变,因此本研究所得结论仍需动物、细胞实验进一步验证。
作者贡献:柴晏、赵玉青、郭旭男负责课题设计、文章撰写;柴晏、郭旭男负责数据库检索及数据整理;赵玉青、王东英负责实验实施及实验数据处理;边云飞负责文章的质量控制及审校。
本文无利益冲突。
猜你喜欢
成都信息工程大学学报(2022年3期)2022-07-21
中学生数理化(高中版.高考理化)(2022年5期)2022-06-01
昆明医科大学学报(2022年1期)2022-02-28
中老年保健(2021年9期)2021-08-24
昆明医科大学学报(2021年8期)2021-08-13
财经(2017年2期)2017-03-10
财经(2016年15期)2016-06-03
财经(2016年3期)2016-03-07
财经(2016年6期)2016-02-24
中国卫生标准管理(2015年25期)2016-01-14
