
血红素加氧酶-1促进巨噬细胞极化的研究进展①
2023-01-16 12:08:34韦佳妮应燕萍赵慧函广西医科大学第一附属医院南宁530021
中国免疫学杂志 2022年19期
韦佳妮 应燕萍 赵慧函 (广西医科大学第一附属医院,南宁 530021)
血红素加氧酶-1(heme oxygenase-1,HO-1)及其产物能在各种病理性应激状态下发挥重要的抗氧化应激、抗炎症损伤、调节细胞凋亡以及血管生成等作用[1]。巨噬细胞是一种高度异质性和可塑性细胞,当机体环境改变时,可在不同刺激作用下发生极化。即使巨噬细胞采用了一种表型,但它仍然保持着对新环境影响做出持续改变的能力[2]。因此,巨噬细胞极化的可逆性具有重要的治疗价值。而近年研究发现,上调单核巨噬细胞中HO-1 表达可使其从促炎作用转变为抗炎作用[3-4]。本文综述了HO-1的调控机制及巨噬细胞的极化,并着重讨论了HO-1在促进巨噬细胞向M2极化的机制作用及其在多种疾病发生发展过程中的作用,以期为多种疾病治疗新思路提供理论基础。
1 HO-1表达的调控机制
HO-1 是一种在细胞应激过程具有细胞保护作用的关键因子,而HO-1 基因的调控涉及红系衍生核因子相关因子2(nuclear factor E2-related factor 2,Nrf2)Nrf2/BTB-CNC 异体同源体1(BTB and CNC ho‐mology 1,Bach1)Bach1/HO-1、NF-κB/iNOS 等细胞内信号转导通路,是一个极其复杂的过程[5]。在Nrf2/Bach1/HO-1 通路中,HO-1 表达受Nrf2 及其阻遏物的调控,Nrf2 正调控HO-1 表达,Bach1 负调控HO-1表达,Nrf2与Bach1在细胞核内的动态平衡关系影响HO-1等抗氧化酶的基因转录[6-7]。Nrf2 是一种调节抗氧化、抗炎基因表达的细胞保护因子,其负调控因子Kelch 样环氧氯丙烷相关蛋白-1(Kelch-like ECH-associated protein-1,keap1)是Nrf2 在细胞质中的结合蛋白[8]。在生理状态下,Nrf2 的Neh2 结构域与Keap1 的DGR 区结合后位于细胞质中,处于抑制状态,当受到胞外氧化应激信号刺激后,Nrf2 与keap-1 解离,转移到细胞核,进而与抗氧化物元件(anti-inflammatory response element,ARE)结合并诱导其下游抗氧化酶如:HO-1 等高表达,可使机体免受毒性物质侵害[9-10]。因此,调节Nrf2核易位并增加HO-1的活性可适当地抑制炎症反应的发生。
2 巨噬细胞极化
巨噬细胞是天然免疫的重要组成部分,在人体正常发育、组织修复及病原体的免疫反应中起重要作用,其功能动态变化的过程称为极化。巨噬细胞处于无刺激或静息时为M0 型巨噬细胞,当受到激活后可具有M1、M2表型。巨噬细胞的经典M1表型和替代M2 表型激活反映了T 细胞的Th1-Th2 极化,代表巨噬细胞激活动态变化状态的两个极端[11-12]。其中,M1 型巨噬细胞主要由IFN-γ、脂多糖(lipo‐polysaccharides,LPS)诱导,通过激活Toll 样受体(Toll-like receptor,TLR)及IFN通路极化,分泌TNF-α、IL-6、IL-1β 等促炎细胞因子,表达CD80、诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)、环氧化酶2(cyclooxygenase-2,COX2)等,能够增强抗原提呈能力和杀菌能力,调节并促进Th1 型免疫应答[13-14]。M2 型巨噬细胞主要由巨噬细胞集落刺激因子(macrophagecolony-stimulating factor,MCSF)、IL-4、IL-13 等刺激活化因子,分泌抗炎细胞因子IL-10、转化生长因子-β(transforming growth factor-β,TGF-β)、白介素1 受体拮抗剂(IL-1RA)、表达精氨酸酶1(arginine-1,Arg-1)、CD206,抑制T 细胞的增殖和活化,调节Th2 型免疫应答[15-16]。M2 巨噬细胞可进一步分为4 个不同的亚群,包括M2a、M2b、M2c和M2d。其中,M2a 亚群由IL-4 和IL-13 诱导,产生高水平的CD206、IL-1RA。M2b 表型可通过免疫复合物和TLR 激动剂或IL-1 受体配体刺激诱导,产生抗炎和促炎细胞因子。M2c细胞具有较强的抗炎活性,可被糖皮质激素和IL-10诱导,释放大量IL-10及TGF-β。M2d 型巨噬细胞可由腺苷或IL-6 诱导极化,IL-10 和血管内皮生长因子的产生增加,TNF-α和IL-12的表达降低[17-18]。
3 HO-1诱导促进巨噬细胞向M2表型极化的分子机制
HO-1 的表达调控及巨噬细胞极化均受微环境变化刺激、细胞因子和信号级联反应介导。诱导HO-1 表达可以抑制NF-κB/MAPK 通路,减少M1 巨噬细胞的极化,激活IL-10/STAT3 和IL-4/STAT6,上调PPAR-γ表达;HO-1还可以上调Nrf2,通过免疫细胞PI3K/Akt通路促进M2巨噬细胞极化。HO-1促进巨噬细胞极化的分子机制如图1所示。
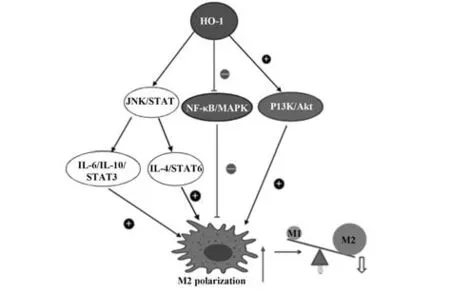
图1 HO-1诱导促进巨噬细胞M2表型极化的分子机制图Fig.1 Molecular mechanism of HO-1-induced M2 pheno⁃typic polarization of macrophages
3.1 Janus 激酶(JAK)/ 信号转导及转录活化因子(signal trans-duction and activators of transcription,STAT)信号通路 JAK/STAT 信号通路是重要细胞因子信号转导通路,参与机体免疫失调和肿瘤发生等过程。JAK由JAK1、JAK2、TYK2和JAK3组成,是一种非受体胞质酪氨酸激酶,可催化磷酸基团向底物蛋白的转移[19]。STAT 由7 个不同的基因编码,分别是:STAT1、STAT2、STAT3、STAT4、STAT5A、STAT5B 和STAT6。其中,STAT3 是JAK 的主要信号转导子之一,在IL-6 的作用下,JAK 被带入糖蛋白130(gp130)近端,进而使STATs磷酸化[20]。IL-6的表达受HO-1负调控,IL-6诱导细胞中STAT3 磷酸化以及HO-1 的基因表达,通过JAK/STAT3 信号传导途径诱导人HO-1 基因表达[21]。有研究报道草属植物中的黄酮类化合物部分通过抑制IL-6/STAT3 信号通路活化,从而影响HO-1的表达,抑制巨噬细胞M2表型极化[22]。巨噬细胞是IL-10 的潜在来源,IL-10和HO-1 通过正反馈环相互联系,IL-10 刺激可促进STAT3 磷酸化和抗炎基因上调,能使巨噬细胞向抗炎M2 表型转变[23]。IL-10/STAT3 和IL4/STAT6 分别是M2c 和M2a 表型极化的两条重要信号通路。STAT6 是调控M2 巨噬细胞极化的关键靶点,STAT6信号通过IL-4 及其各膜受体IL-4Rα 结合而被激活,这导致JAK2 的信号转导和STAT6 磷酸化,然后使其二聚化,磷酸化的STAT6 易位至细胞核并诱导基因转录[24]。作为HO-1 的强诱导剂,血红素对HO-1的诱导可促进IL-4 和STAT6 磷酸化,通过STAT6 磷酸化的IL-4信号诱导过氧化物酶体增殖物激活受体γ(peroxisome proliferators-activated receptors,PPAR-γ)及其激活因子的转录,增强M2 表型巨噬细胞的表达[25]。
3.2 NF-κB/MAPK 信号通路 NF-κB 是一种B 细胞特异性转录因子,组成包括RelA(p65),RELA、RELB、REL、p50 和p52[26]。在非刺激状态下,NF-κB在细胞质中与抑制蛋白IκB 结合,保持静息状态。当受到刺激时,抑制蛋白IκB 被IκB 激酶磷酸化和降解,NF-κB被释放并转移到细胞核,与靶基因的启动子区域对应点相结合、调控下游相关基因转录并触发促炎递质释放[27]。NF-κB途径活化与丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)如细胞外调节激酶(ERK),c-Jun N 端激酶(JNK)和p38的磷酸化有关,MAPK 在调节巨噬细胞诱导炎症基因表达的炎症分子和免疫相关的细胞毒性因子中起重要作用[28]。氧化应激信号通过激活NF-κB/MAPK信号通路在增强炎症中发挥重要作用。而Nrf2 和NF-κB 是调节细胞对氧化应激和炎症反应的两个关键转录因子。研究表明,这两种重要途径之间存在功能上的相互作用[29]。NF-κB通过拮抗Nrf2下调HO-1表达,HO-1受NF-κB调控,可抑制NF-κB介导的促炎基因表达[30]。因此,HO-1诱导也被证明通过 NF-κB 的失活抑制促炎症递质表达。大量研究表明,天然化合物调节巨噬细胞极化可作为一种有前景的治疗策略。稻瘟散(OR)对LPS 刺激的RAW264.7 细胞中炎症细胞因子的产生以及iNOS 和COX-2 的表达具有强烈的抑制作用。这些作用归因于通过抑制IκBα降解、阻断MAPK 磷酸化及影响HO-1 表达抑制NF-κB 活化[31]。一项研究结果表明MH 乙醇提取物(MHE)通过抑制经由NF-κB 的炎症递质产生和MAPK 信号通路抑制和诱导巨噬细胞HO-1表达而发挥抗炎作用[32]。
3.3 磷酸酰肌醇-3-激酶(PI3K)/蛋白激酶B(AKt)信号通路 巨噬细胞激活是一种严格调控的现象,由受体刺激或细胞内调控蛋白引发的多个信号级联反应决定。在不同的信号级联反应中,PI3K/Akt途径及其下游靶点已成为巨噬细胞活化表型的中央调节因子[33]。Akt 是一种丝氨酸/苏氨酸激酶,被PI3K 激活。PI3K/Akt 信号通路的激活可以调节调节巨噬细胞的分化,迁移和增殖,也可协调巨噬细胞对不同代谢和炎症信号的反应,低水平的Akt 信号传导诱导炎症M1表型,高水平的Akt活性诱导替代性M2 巨噬细胞[34-35]。先前的研究表明,HO-1 基因表达可通过免疫细胞中的PI3K/Akt 通路上调,可在氧化应激损伤中提供抗凋亡或抗炎作用,抑制PI3K/Akt 激活可抑制HO-1 的表达[36]。PI3K/Akt 通路的激活对于限制TLR 刺激的巨噬细胞的促炎反应和促进抗炎反应至关重要,并且被认为是巨噬细胞中TLR和NF-κB信号的负调节剂[37]。PI3K/Akt途径在LPS诱导的巨噬细胞中表现出抗炎作用可作为抗炎药物的潜在靶点。在LPS 激活的巨噬细胞中,α7 烟碱乙酰胆碱受体(α7nAChR)通过蛋白激酶A的激活介导HO-1 的诱导,并刺激PI3K 的活性。由此产生的Nrf2 核易位诱导HO-1 表达[38]。因此,PI3K/Akt 可作为上游信号分子激活Nrf2,诱导HO-1表达并促进巨噬细胞向M2 表型极化,从而增强细胞防御炎症和氧化损伤的防御系统。
4 HO-1 表达调节巨噬细胞极化,参与多种疾病的发生发展
由于HO-1 具有细胞类型特异性的免疫调节作用,靶向上调髓系细胞中HO-1 的表达可能是一种直接的抗炎治疗。越来越多的证据表明,特异性诱导HO-1 可以减轻炎症反应。当心、肝、肺、肾等脏器损伤时,HO-1 表达调节可使巨噬细胞向M2 表型极化,发挥其组织保护作用。研究发现,将含HO-1基因的腺病毒转染巨噬细胞(Ad-HO-1-Mφ)移植到大鼠急性心肌梗死区后仍能高表达HO-1 蛋白并显著减轻梗死区的炎症和氧化应激水平[39]。骨髓特异性HO-1 基因表达驱动巨噬细胞向M2 抗炎表型极化并保护肝脏缺血灌注再损伤(ischemia-reperfu‐sion injury,IRI),其缺乏会促进M1 型极化,加剧肝脏IRI[40]。野生型小窝蛋白-1 脚手架区结构域(CSD)多肽可增加HO-1 活性并促进肺泡巨噬细胞(alveolar macrophage,AM)由M1 表型向M2 表型转化,抑制促炎因子表达,可治疗LPS引起的急性肺损伤(acute lung injury,ALI)[41]。在急性肾损伤(acute kidney injury,AKI)和进行性慢性肾脏病中可在肾小球及肾间质中检测到巨噬细胞浸润,且巨噬细胞可根据肾脏微环境变化,在促炎表型和抗炎表型之间进行转化从而影响肾脏损伤疾病转归[42]。一些感染性疾病如哮喘、慢性阻塞性肺疾病、寄生虫感染等,在感染期间,常驻组织巨噬细胞和炎症单核细胞从血液中并分化为巨噬细胞,诱导聚集炎症以促进机体清除病原体。巨噬细胞在许多慢性炎症和自身免疫性疾病的发病机制中发挥重要作用。其中,M1 巨噬细胞中的许多炎症细胞因子,已被确定为慢性炎症和自身免疫性疾病的重要介质和驱动因素[43]。近十年的研究表明,巨噬细胞具有促肿瘤功能,与肿瘤进展密切相关[44]。肿瘤相关巨噬细胞(tumor-associated macrophages,TAMs)大量浸润大多数实体肿瘤,可通过刺激增殖、血管生成、转移和提供抗肿瘤免疫屏障而促进肿瘤进展[45]。人类肿瘤样本的研究显示,在许多恶性肿瘤中,M2 表型的巨噬细胞密度较高,且与较差的临床预后密切相关。据报道,利用免疫细胞对抗肿瘤的癌症免疫疗法被证明是抗击癌症的有力武器,浸润性TAMs 本身或TAMs 极化通路被认为是治疗恶性肿瘤的新靶点,并越来越多地应用于临床[46]。从自身免疫到心血管病理,从感染性疾病到癌症,巨噬细胞都是广泛的疾病中的主要参与者。因此,研究巨噬细胞极化的可逆性可为多种疾病的干预和治疗提供新思路。
5 小结与展望
巨噬细胞在免疫系统中发挥着关键作用,在一定的稳态和病理条件下经历一系列功能激活状态。巨噬细胞具有高度的异质性和可塑性,其表型和功能可在不同微环境中进行动态转换,既可以通过产生促炎细胞因子的Th1 反应促进炎症(M1 表型巨噬细胞),也可以通过产生抗炎细胞因子的Th2反应抑制炎症(M2 表型巨噬细胞)。巨噬细胞在多种疾病中既有保护作用,又有致病作用。共同证据表明,巨噬细胞在局部环境中分化、极化、复极化和活化的变化在多种自身免疫性和炎症性疾病的发病机制中起决定性作用。当感染或炎症严重时,巨噬细胞首先表现为M1 表型,针对刺激释放TNF-α 等促炎介质,但如果M1 期持续,可能会造成组织损伤。此时,M2巨噬细胞分泌抑炎因子,抑制炎症反应,促进组织修复、重塑、血管生成和维持体内平衡。HO-1属于巨噬细胞表达的抗炎基因,HO-1上调能促进巨噬细胞向M2 表型极化,发挥抗炎作用,参与多种疾病的发生发展。说明靶向诱导HO-1 可能在临床上具有极大的治疗潜力,HO-1介导的巨噬细胞极化治疗策略对治疗疾病具有可能性,但还需要在临床中进行更深入的研究。
猜你喜欢
现代财经-天津财经大学学报(2022年5期)2022-06-01 06:08:32
中华养生保健(2020年7期)2020-11-16 01:13:32
现代园艺(2017年21期)2018-01-03 06:41:32
中成药(2017年9期)2017-12-19 13:34:20
电子测试(2017年15期)2017-12-18 07:18:51
中成药(2017年10期)2017-11-16 00:50:09
中国康复理论与实践(2015年10期)2015-12-24 05:42:44
中药与临床(2015年5期)2015-12-17 02:39:30
电源技术(2015年1期)2015-08-22 11:16:18
医学研究杂志(2015年5期)2015-06-10 06:43:26
