
FoxO1/p38 MAPK信号通路在LPS致急性肺损伤中的作用研究
2023-01-05 10:17:12杨亚丽王艳佳杨晓玲
中国药理学通报 2023年1期
杨亚丽,田 荣,袁 茵,王艳佳,杨晓玲
(1.宁夏医科大学基础医学院;2.国家卫生健康委员会代谢性心血管疾病研究重点实验室;3.宁夏血管损伤与修复研究重点实验室,宁夏 银川 750004)
急性肺损伤(acute lung injury, ALI)是呼吸系统一种严重的疾病,其死亡率高,发病率约为30%~40%[1]。越来越多的证据表明,过度炎症反应在ALI的发病中起关键作用[2-3]。作为革兰阴性细菌细胞壁主要成分的脂多糖(lipopolysaccharide, LPS),体内外实验均表明,LPS诱导ALI是一种被广泛接受的细菌性脓毒症动物模型[4]。FoxO1作为Fox转录家族的主要成员之一,可以调节多种基因的表达,参与细胞分化、氧化应激、炎症反应、脂代谢等病理过程,并参与ALI疾病发生[5-6]。丝裂原活化蛋白激酶(mitogen activated protein kinases,MAPKs)信号通路是细胞内调控炎症产生的主要信号转导通路,与LPS诱发的ALI有密切关系[7],p38 MAPK通过磷酸化被激活后,调控下游转录因子,促进IL-1、TNF-α等细胞因子的分泌,诱导一系列炎症反应[7]。因此,本研究通过LPS复制ALI体内外模型,探讨FoxO1 DNA甲基化及FoxO1/p38 MAPK信号通路在ALI中的作用及意义。
1 材料与方法
1.1 材料
1.1.1实验动物 20只SPF级CBS+/+♂小鼠饲养于宁夏医科大学实验动物中心标准屏障环境内,通风良好,期间自由活动和饮食,饲养8周后,体质量约25 g,实验动物合格证号:SCXK(京)2016-0006,根据实验安排和需要,对小鼠进行处理。
1.1.3仪器 超净工作台(苏州,安泰);CO2培养箱(德国,Heraeus);5415D型微量台式离心机(德国,Eppendorf);BS110S型精密天平(德国,Sartorius);荧光定量PCR仪(美国,Bio-Rad);垂直电泳仪、凝胶成像仪和Mod-el680全自动酶标仪(美国,Bio-Rad),酶标仪(美国Bio-TEK),普通PCR仪和qRT-PCR仪(德国,耶拿)。
1.2 方法
1.2.1动物分组 选取20只8周龄25 g左右的SPF级CBS+/+小鼠,随机分为LPS组和Control组,每组10只,饲喂普通饮食,参考文献说明[8],术前将LPS溶解于无菌生理盐水中,小鼠禁食12 h后,LPS组根据5 mg·kg-1的剂量给予LPS腹腔注射,Control组给予等量的生理盐水腹腔注射。造模成功6 h后麻醉处理动物获取肺组织。本研究严格按照《实验动物管理条例》进行实验。
1.2.2细胞培养 PVEC 使用含10%胎牛血清及1%的青链霉素的DMEM培养液,置于37 ℃、5% CO2的恒温培养箱中常规培养,隔日换液,根据细胞生长状态,传至2~3代可用于后续实验。
1.2.3细胞分组及转染 实验组:PVEC转染 si-FoxO1 组;阴性对照组(negative control,si-NC):转染对基因无影响的空载RNA;空白对照组(control):加入等体积的完全培养基组。将处于指数生长期的细胞消化后细胞计数,等量接种于6孔板,待细胞生长到肉眼观60%~70%密度时进行转染。依据Lipofectamine2000 Reagent和si-FoxO1的说明书对各组细胞进行细胞转染实验,培养6 h后,换正常的培养基,48 h后进行后续实验。
1.2.4HE染色 处死小鼠后,取左肺上叶组织置于4%的多聚甲醛固定24 h,进行常规脱水、石蜡包埋、切片,按标准操作进行HE染色,光学显微镜下观察肺组织的病理学变化。
1.2.5免疫组化染色 使用上述的方法制备的石蜡切片,经常规脱蜡水化,于0.01 mol·L-1柠檬酸盐溶液高温加热进行抗原修复2 min后,每张切片滴入0.3%过氧化氢以阻断内源性过氧化物酶的活性。经置室温冷却后,滴入10%山羊血清37 ℃封闭30 min,以减少非特异性染色。滴加抗FoxO1(1 ∶200)单克隆抗体在湿盒中4 ℃孵育过夜,次日滴加生物素标记的二抗,室温孵育1 h,加入DAB显色,再使用苏木精染色液对切片进行复染,常规脱水中性树脂封片,置于显微镜下观察并拍照。
1.2.6肺组织炎症因子的检测 各组小鼠处死后迅速取肺组织,称取约50 mg加入450 μL PBS,匀浆器充分研磨组织直至完全裂解,12 000×g离心10 min,取上清,按照ELISA说明书,检测肺组织中IL-6和TNF-α的含量。
青海省平均海拔3500~4500m,是我国生态安全、水资源利用和高寒生物多样性保护的关键地区。青海省现有可造成土壤污染的企业有矿业、铬化工企业、铝镁化工企业、石油企业等多家企业,都对土壤环境造成威胁。
1.2.7Western blot检测FoxO1、DNA甲基转移酶和p38 MAPK的蛋白表达 称取Control组及LPS组肺组织各0.6 g,用预冷的PBS冲洗组织2次,加入细胞裂解液,使用匀浆机将其破碎,在4 ℃摇床裂解组织2 h,离心机4 ℃,12 000×g离心5 min,提取上清液,即得总蛋白,采用BCA法测定各组蛋白含量,按每孔30 μg蛋白经煮沸8 min后,开始10% SDS-PAGE凝胶电泳,随后将蛋白质湿转到PVDF膜上。用5%脱脂奶粉室温封闭2 h。加入一抗(1 ∶1 000)孵育,4 ℃摇床过夜,次日PBST洗膜,10 min,洗3 次,然后使用含HRP-二抗室温孵育2 h,PBST洗膜3次后将PVDF膜放入凝胶成像仪内,膜上滴ECL发光液,A液 ∶B液=1 ∶1,曝光,以β-actin为内参,Image Lab图像分析软件分析各组蛋白的相对表达。
1.2.8qRT-PCR检测FoxO1和DNA甲基转移酶的mRNA水平 称取各组组织约0.6 g,使用匀浆机将其破碎,加1 mL TRIzol RNA提取试剂,提取总RNA,测定样品纯度(A260/A280=1.80~2.0)与浓度后将mRNA逆转录为cDNA。将得到的cDNA按照Bestar SybrGreen qPCR Master Mix 10 μL、上游引物0.8 μL、下游引物0.8 μL、cDNA 2 μL、ROX 0.4 μL,灭菌蒸馏水6.0 μL,配成20 μL的PCR扩增体系。在NCBI查询各基因的CDS,用Premier 6.0软件设计引物。引物序列见Tab 1。扩增条件如下:95 ℃预变性2 min,95 ℃变性10 s,57 ℃退火30 s,72 ℃延伸30 s,40个循环,以GAPDH作为内参,目的基因的相对表达用2-ΔΔCT法分析。

Tab 1 Primers involved in real time PCR
1.2.9nMS-PCR检测FoxO1启动子区DNA甲基化水平 按DNA提取试剂盒说明书提取各组细胞全基因组DNA,亚硫酸盐修饰法对全基因组DNA进行甲基化修饰。nMS-PCR法检测FoxO1启动子区DNA甲基化程度的改变。针对FoxO1启动子区,在线(http://www.urogene.org/cgi-bin/methprimer/methprimer.cgi)设计一对外引物及两对内引物(外引物:上游:5′-TTTGTAGGTGTGTATAGG TAGGGTG-3′,下游:5′-AATACTCCAAACAAAACCCAAAC-3′;甲基化内引物:上游:5′-TAGAAAAGCGGTTTTTATAGAAGAC-3′,下游:5′- TACCTATACACACCTACAAAACGAA-3′;非甲基化内引物:上游:5′-TGGTAGAAAAGTGGTTTTTATAGAAGAT-3′,下游:5′-CCCTACCTATACACACCTACAAAACA-3′)。反应体系:MIX 12.5 μL、H2O 7 μL、上下游引物各1 μL、已修饰的DNA模板3.5 μL,共25 μL。外引物扩增的反应条件为:95 ℃ 2 min;95 ℃ 20 s,60 ℃ 30 s,72 ℃ 30 s,20个循环,每个循环降0.5 ℃至50 ℃,72 ℃ 2 min。以外引物的PCR产物为模板,进行内外引物的扩增,反应条件同外引物。随后,取10 μL PCR产物于2%的琼脂糖凝胶上电泳,用凝胶成像分析仪成像并分析甲基化条带及甲基化条带的光密度,按如下公式进行计算:甲基化/%=甲基化OD值/(甲基化OD值+非甲基化OD值)×100%。

2 结果
2.1 肺组织的形态学变化正常肺脏组织壁薄,无结缔组织增生、增厚;各级支气管结构完整清晰,染色较为均匀,未见明显变性坏死(Fig 1A);LPS 组小鼠肺泡结构被破坏,肺泡壁溶解断裂,局部肺泡腔狭窄甚至消失,肺泡壁、肺泡腔或间质炎细胞大量浸润(Fig 1B)。

Fig 1 Lung pathological changes of mice in each group (200×)
2.2 各组小鼠IL-6和TNF-α的表达与Control 相比,LPS组小鼠肺组织IL-6和TNF-α浓度及mRNA水平均明显升高,其中ELISA结果显示,IL-6和TNF-α浓度分别升高了38.9%和52.5%(Fig 2A,P<0.05),qRT-PCR结果显示,IL-6和TNF-α的mRNA水平分别升高了144.5%和246.9%(Fig 2B,P<0.01),且差异有统计学意义。
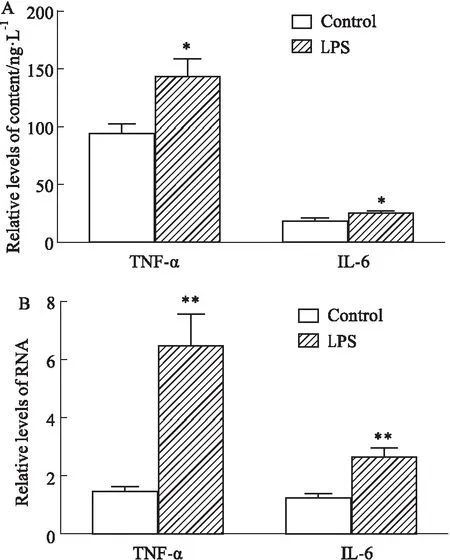
Fig 2 Expression of inflammatory cytokines IL-6
2.3 肺组织中FoxO1的表达变化肺组织免疫组化分析显示:与Control组比较,LPS组FoxO1的表达高于正常肺组织的表达(Fig 3A);qRT-PCR和Western blot分别检测肺组织中FoxO1 mRNA及蛋白的表达水平,结果显示:与Control组比较,LPS组FoxO1的mRNA和蛋白表达分别增加了134.9%和61.8%(Fig 3B和C),且差异有统计学意义(P<0.01)。

Fig 3 Expression of FoxO1 in lung tissues of
2.4 FoxO1 DNA 甲基化水平改变利用生物信息学软件分析预测FoxO1启动子区结构,在启动子上游-1226~-1352和-1414~-1796 bp的位置存在两个CpG岛(Fig 4A)。利用nMSP 检测FoxO1启动子区的改变,结果显示,与对照组相比,LPS组FoxO1启动子区甲基化水平降低了17.2%(Fig 4B,P<0.05)。
DNA甲基转移酶是维持DNA甲基化修饰的酶,主要包括DNMT1、DNMT3A和DNMT3B,qRT-PCR及Western blot分别检测了DNMT1、DNMT3A、DNMT3B的表达,结果显示,LPS组小鼠肺组织DNMT3A和DNMT1的mRNA水平分别降低了149.1%和59.8%(P<0.05,P<0.01),DNMT3B变化差异无统计学意义(P>0.05,Fig 4C);DNMT3A和DNMT3B的蛋白水平分别降低了142.1%和37.7%(P<0.05,P<0.01),DNMT1的变化无统计学意义(P>0.05,Fig 4D),综合以上数据,说明FoxO1启动子区发生甲基化主要受到DNMT3A的调控,从而参与LPS导致的肺损伤过程。
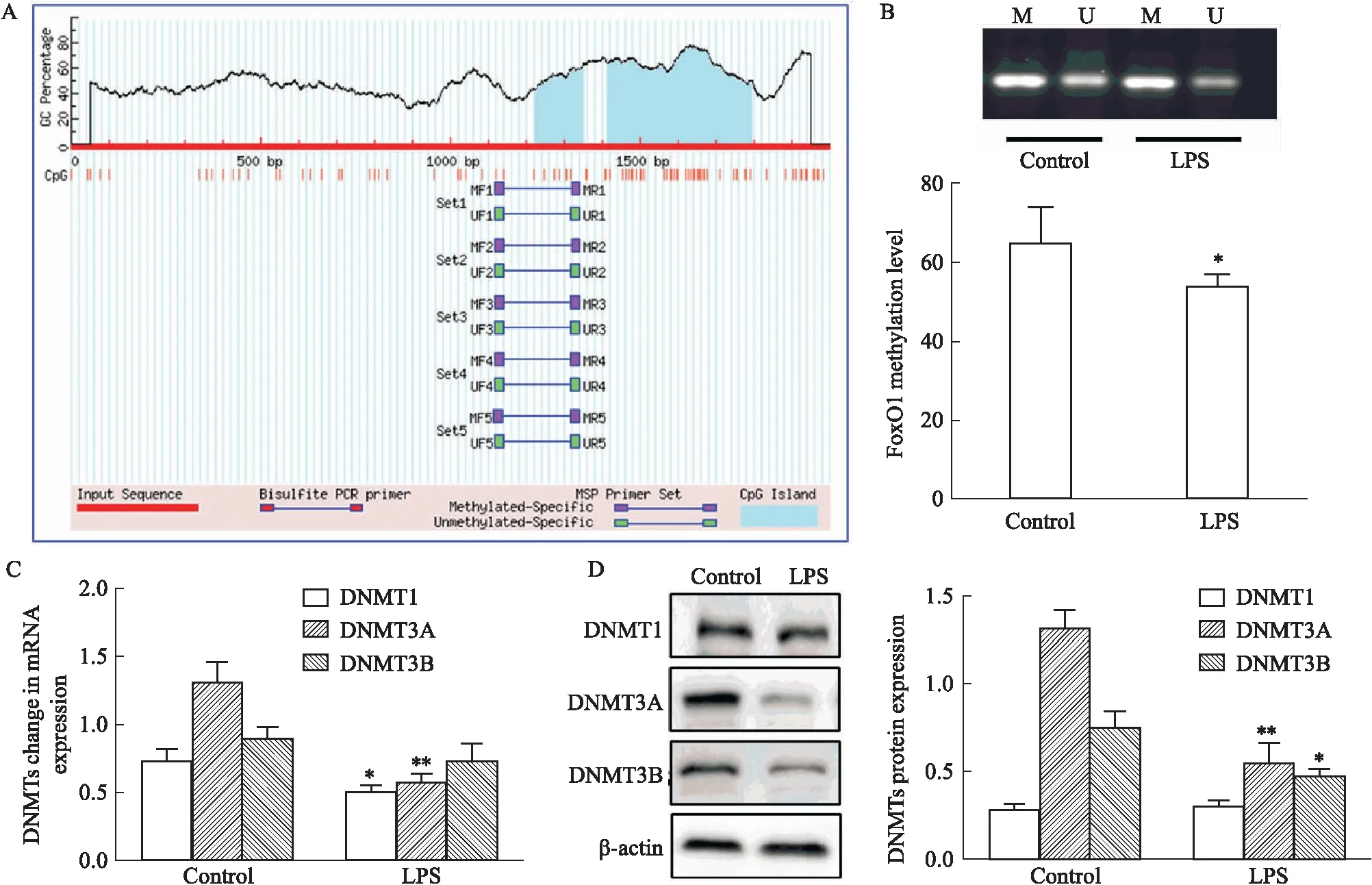
Fig 4 Expression of DNA methyltransferase in lung tissues
2.5 FoxO1甲基化水平与炎症的相关性分析Pearson相关性分析结果显示,FoxO1甲基化水平与小鼠肺组织IL-6及TNF-α浓度呈负相关(r2=-0.521 8,r2=-0.788 2)(Fig 5A,B,P<0.05,P<0.01),该相关性分析表明,小鼠肺组织炎症因子的产生与FoxO1启动子区DNA甲基化水平相关。

Fig 5 Analysis of correlation between FoxO1
2.6 p38 MAPK的磷酸化水平与Control对照比较,LPS组肺组织p38 MAPK蛋白表达无差异(P>0.05),而p38 MAPK磷酸化水平明显增加,升高了134.1%(P<0.01),见Fig 6。
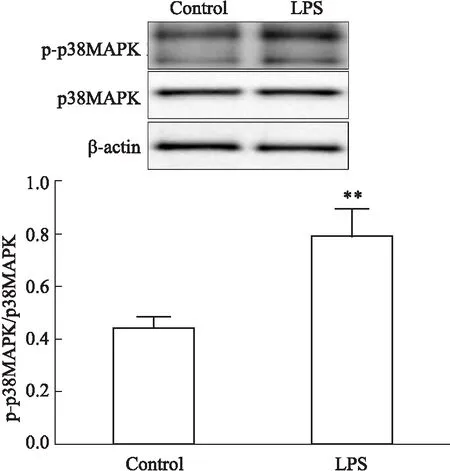
Fig 6 The phosphorylation level of p38
2.7 干扰FoxO1后p38 MAPK的表达变化为了进一步探讨FoxO1在肺损伤中的机制,构建FoxO1干扰片段并转染至 PVEC 48 h,qRT-PCR结果显示,在引起FoxO1表达下调的3个干扰片段中,FoxO1-siRNA-1803( si-FoxO1)敲低效果最佳,可用于后续试验(Fig 7A)。Western blot验证其干扰效率,结果显示,FoxO1干扰片段筛选成功(Fig 7B);为证明FoxO1在LPS诱导的肺损伤中调控作用,在LPS干预PVEC情况下转染si-FoxO1,Western blot检测p38 MAPK的磷酸化变化。结果显示,与LPS+si-NC组相比,LPS+si-FoxO1组p38 MAPK蛋白磷酸化水平明显降低(Fig 7C),以上结果说明,FoxO1可以调控p38 MAPK的磷酸化水平。
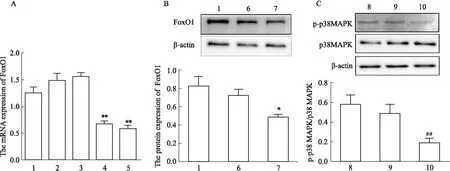
Fig 7 Changes of p38 phosphorylation level after interference with
3 讨论
脂多糖是革兰阴性菌细胞壁的主要活性成分,可诱导炎症反应的过度激活,现已被广泛应用于诱导ALI模型[9-10]。我们的实验给予小鼠腹腔注射LPS诱导构建了急性肺损伤小鼠模型,肺损伤小鼠模型中病理学表现为肺泡结构损伤,肺间质充血水肿、大量炎性细胞浸润,肺组织匀浆液TNF-α和IL-6含量也明显高于对照组,说明我们成功构建了急性肺损伤小鼠模型。
FoxO1作为人体最重要的转录因子之一,在多种细胞类型中广泛表达,可通过转录和翻译,调节细胞氧化应激反应、炎症及增殖等多种病理生理过程[11]。FoxO1自身的转录后修饰调节FoxO1的功能奠定了它在代谢、免疫等方面研究中的重要地位[12]。Artham等[13]发现,Akt1/FoxO1/stromelysin1通路参与了LPS诱导的ALI的发病机制中,而抑制FoxO1的表达后,可逆转脂多糖诱导的急性肺损伤。本研究结果显示:在LPS诱导的肺损伤模型中,FoxO1表达明显增加,表明FoxO1参与了肺损伤的过程,但其具体机制有待进一步研究。
DNA甲基化作为真核生物最常见的表观遗传修饰方式之一,目前,FoxO1 DNA甲基化作为一种调控机制日益成为人们研究的热点,课题组前期发现,FoxO1启动子区的低甲基化造成FoxO1的表达增加,在Hcy诱导的肝细胞内质网应激未折叠蛋白反应中发挥重要作用[14],但是,关于FoxO1基因DNA异常甲基化与肺损伤的相关性研究尚属于全新的领域,其具体作用尚不明确。我们通过生物信息学分析发现,FoxO1启动子区存在两个CpG岛,提示FoxO1的表达可能受甲基化调控。进一步用MSP检测FoxO1启动子区甲基化,结果显示,LPS组肺组织FoxO1启动子区甲基化水平明显降低,与其表达水平相反。由于DNA甲基化受甲基转移酶调控,进一步检测甲基转移酶的表达,发现LPS组DNMT3A表达明显降低,而DNMT1和DNMT3B无明显变化,提示DNMT3A表达降低使FoxO1启动子区发生低甲基化,进而调控FoxO1的表达。
p38MAPK作为MAPKs家族重要信号转导通路之一,在机体内被促炎细胞因子、内毒素等因素诱发后,介导机体的炎症、应激和损伤等信号传递过程,并且活化p38 MAPK信号通路,以磷酸化形式为主要特征[15]。Viji等[16]报道,高同型半胱氨酸血症通过扰乱FoxO1和MAPK信号级联反应来改变成骨细胞中的氧化还原调节机制。此外,亦有研究发现[17],在肝细胞中,FoxO1通过增加p38活性,进而激活PKB和MAPK通路。提示FoxO1/p38 MAPK信号通路在多种生理和病理过程中起重要作用。本研究也发现,模型组肺组织p38 MAPK全蛋白无明显变化,但其磷酸化水平明显增加,为了进一步明确FoxO1 DNA甲基化水平与p38 MAPK磷酸化之间的关系,我们继续体外培养小鼠肺血管内皮细胞,并转染si-FoxO1干扰片段检测p38 MAPK的磷酸化水平,发现其磷酸化水平明显降低,提示脂多糖促使p38 MAPK信号通路活化,并参与调控小鼠肺损伤。
综上所述,LPS可诱导小鼠肺组织发生炎症反应,并通过激活相关的信号通路导致肺损伤,其作用机制可能与FoxO1启动子区DNA甲基化水平降低,进而上调FoxO1的表达并激活p38 MAPK信号通路有关。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04 11:40:51
生物学通报(2022年1期)2022-11-22 08:12:18
南京林业大学学报(自然科学版)(2021年5期)2021-10-13 02:06:16
天津医科大学学报(2019年6期)2019-08-13 07:04:42
广西林业科学(2016年3期)2016-03-16 05:43:25
安徽医科大学学报(2015年9期)2015-12-16 11:09:42
现代检验医学杂志(2015年2期)2015-02-06 02:00:48
沈阳医学院学报(2014年4期)2014-12-27 13:44:30
遗传(2014年3期)2014-02-28 20:59:01
遗传(2014年3期)2014-02-28 20:58:49
