
葡萄籽原花青素低聚体抑制A1型星形胶质细胞极化机制
2023-01-05 12:13杨智超董艺薇苑舒文李彦青宋丽娟黄建军马存根
中国药理学通报 2023年1期
王 青,杨智超,董艺薇,苑舒文,李彦青,宋丽娟,黄建军,马存根,3
(1. 山西中医药大学神经生物学研究中心,国家中医药管理局益气活血法治疗多发性硬化重点研究室, 山西 晋中 030619;2. 国药同煤总医院神经外科/山西省卫健委神经疾病防治研究重点实验室,山西 大同 037003;3. 山西大同大学脑科学研究所,山西 大同 037009)
中枢神经炎性脱髓鞘疾病是一类以炎性反应和广泛原发性髓鞘脱失为主要特征的疾病。其中复发-缓解型的多发性硬化(multiple sclerosis,MS)、视神经脊髓炎谱系疾病(neuromyelitis optica spectrum disorders,NMOSD)是我国最常见的脱髓鞘疾病,病程通常表现为复发-缓解交替进行,多次发作造成神经功能不可逆的损伤,是中青年非创伤性致残的主要原因之一[1-2]。长期以来,减少髓鞘的炎性免疫损伤是治疗的重要靶点,但尚未取得理想的疗效。
星形胶质细胞(astrocytes,AS)是中枢神经系统(central nervous system,CNS)最丰富的胶质细胞,以连续和基本不重叠的方式整齐有序地覆盖整个CNS,可通过参与髓鞘形成、维护血脑屏障、调节突触功能和调节能量代谢以维持CNS的稳态。一旦中枢受损,AS迅速作出反应,大量反应性AS增生,并借由本身的功能和表型的转化对周围的神经元和胶质细胞产生有益或有害的影响。如NMOSD、MS及其动物模型-实验性变态反应性脑脊髓炎(experimental autoimmune encephalomyelitis,EAE)和双环己酮草酰二腙(cuprizone,CPZ)诱导的脱髓鞘模型中均存在着的数量众多的反应性AS[3-4],可通过释放多种抑制性因子如硫酸角质素蛋白聚糖、少突胶质细胞髓鞘蛋白和髓鞘相关蛋白,以及炎性因子如肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、白介素-1α(interleukin-1α,IL-1α)和补体1q(complement 1q,C1q)等损伤神经元和髓鞘细胞[3,5]。而靶向AS可明显缓解髓鞘脱失,如Dalahmah等[6]发现半乳糖凝集素-3(galectin-3,Gal-3)可通过促进AS的增殖和炎性反应加重少突胶质细胞的损伤。因此,随着对反应性AS功能及其作用机制的深入研究,它已逐渐成为保护和促进髓鞘再生的一类重要潜在靶细胞。
葡萄籽原花青素低聚体(grape seed proanthocyanidins,GSPs)是最为安全有效的抗氧化剂之一,具有抗炎、抗恶性肿瘤、免疫调节、抗衰老和神经保护等药理作用,被广泛应用于食品、化妆品、药品和保健品等领域[7-9]。基于其优良的抗炎作用,我们前期探索了GSPs对CPZ脱髓鞘小鼠的治疗作用,发现GSPs可显著缓解CPZ诱导的髓鞘脱失,减少中枢炎性因子的分泌[10],同时诱导胶质纤维酸性蛋白(glial fibrillary acidic protein,GFAP)阳性的AS在病灶区的富集,说明GSPs可能通过AS发挥神经保护作用。因此,本研究以体外原代AS为研究对象,初步探讨了GSPs靶向AS的作用机制。
1 材料与方法
1.1 原代星形胶质细胞的培养将新出生2-7 d的C57BL/6小鼠,用冰低温麻醉2~10 min后,用75的乙醇对幼鼠的头部和颈部进行消毒后进行断头处理。将大脑取出后,置于冰浴的高糖DMEM(Gibco,11995040)中,在体视显微镜下仔细剥离脑膜,用灭菌后的眼科剪将脑组织剪成1 mm3的碎片。将组织碎片置于0.25%胰蛋白酶EDTA溶液(Thermo Fisher Scientific)中,温和摇动培养10 min后,添加完全培养液中止胰蛋白酶反应。完全培养液是含有10%胎牛血清的高糖DMEM。消化的组织以300×g×5 min离心,小心地去除上清液,并重悬于高糖DMEM中,通过40 μm的nylon膜将组织分离成单细胞悬液。获得的单细胞悬液接种于T75培养瓶中,以获得原代混合胶质细胞。培养瓶提前用100 mg·L-1的多聚赖氨酸(分子量为15~30万)包被后在4 ℃过夜,用PBS洗涤3次并在使用前干燥。原代混合胶质细胞培养2 d后更换新鲜的完全培养液,此后每3 d更换1次培养液,通常7~11 d混合胶质细胞会铺满培养瓶底。将长满的培养瓶放置于摇床以180 r·min-1的转速摇动24 h以去除小胶质细胞(microglia,MG)。然后加入新鲜的AS培养基(ScienCell,1801),并以240 r·min-1的转速摇动6 h以去除少突胶质细胞前体细胞(oligodendrocyte progenitor cells,OPCs)。剩下的细胞在实验前差速贴壁30 min,进一步纯化AS。
1.2 免疫荧光染色法待细胞融合度达到80%左右时,移除培养液,PBS洗涤3次后,室温4%多聚甲醛固定10 min,0.3%的TritonX-100通透10 min。牛血清白蛋白工作液室温封闭后,加入抗GFAP抗体(CST,12389),4 ℃孵育过夜。加入FITC标记的二抗,避光室温孵育1.5 h。DAPI避光孵育染核20 min,滴加抗荧光淬灭封片液(Thermo,P36970),封片后使用荧光显微镜检测(Leica,DM40008)。
1.3 细胞活力检测实验原代AS进行实验之前,免疫荧光法染色AS标记物胶质纤维酸性蛋白(GFAP)进行纯度检测,其阳性率超过95%。将原代AS接种于96孔板,培养24 h后,分别用不同浓度(0、2.5、5、10、20、30和50 mg ·L-1)的GSPs孵育24 h。乳酸脱氢酶(LDH)法:将96孔板以300×g离心5 min,每孔吸取上清液置于另一平底96孔板,按照LDH检测试剂盒(Solarbio,BC0685)说明书检测各组上清液中LDH的含量。MTT法:将MTT工作液(5 mg ·L-1)加到96孔板,每孔加入10 μL,在CO2培养箱里孵育4 h,弃去培养液,每孔加入100 μL的DMSO,溶解后在490 nm处检测其光密度optical density(OD)值。
1.4 DPPH、ABTS自由基清除实验DPPH实验:分别将10 μL的空白提取液、阳性标准品溶液和不同浓度的GSPs(0、2.5、5、10、20和30 mg·L-1)加入到190 μL的DPPH工作液中,室温避光孵育30 min,在最大吸收峰515 nm处检测吸光度(OD)值,计算清除率/%=(A空白-A测定)/A空白×100%。ABTS实验:空白组:将24 μL的ddH2O加入到180 μL的ABTS混合液中;GSPs组和阳性对照组维生素C(Vitamin C,Vc):分别将6 μL的阳性标准品溶液和不同浓度的GSPs加入到180 μL的ABTS混合液和18 μl蒸馏水中,反应10 min后,于最大吸收峰593 nm处测定吸光度值,计算清除率/%=(A空白-A测定)/A空白×100%。
1.5 A1型反应性星形胶质细胞模型的建立、药物干预和细胞因子检测原代AS用3 μg·L-1的IL-1α(PeproTech,315-01A)、30 μg·L-1的TNF-α(PeproTech,215-11A)和400 μg·L-1的C1q(MyBioSource,MBS143105)孵育24 h,建立A1型AS。30 μg·L-1的GSPs(购自沐凡生物科技有限公司)加入上述的模型中,孵育24 h以干预AS的极化。实验分组为正常组(Nor)、A1型AS组(Model)和GSPs干预组(GSPs)。24 h后更换新鲜的AS培养液,收集上清采用ELISA法检测相关的细胞因子。使用R&D公司的夹心ELISA试剂盒检测TNF-α、IL-1α、IL-6、IL-17和转化生长因子(Transforming growth factor-β,TGF-β)等细胞因子。使用凡科维公司ELISA试剂盒检测睫状神经营养因子(Ciliary neurotrophic factor,CNTF)、H2O2和ROS等因子。细胞因子单位为ng·L-1。
1.6 RT-PCR法PBS洗涤细胞2次后,收集各组的原代AS,使用RNAiso Plus+(TaKaRa,9108)从细胞中提取总RNA。将提取的总RNA用PrimeScriptTM1st Strand cDNA Synthesis Kit(TaKaRa,6110A)逆转录为第一链cDNA。使用TB Green® Premix Ex TaqTMROX plus(TaKaRa,RR42LR)和CFX96 Optics Module荧光定量PCR仪(Bio-Rad)进行定量PCR扩增。引物序列参考已经发表的文献,由上海BioTNT公司合成[11-12]。采用2-ΔΔCt法计算mRNA表达量。Tab 1是使用的引物序列。

Tab 1 Primers for RT-PCR
1.7 Western blot法移除培养液,用PBS洗涤细胞两次以去除杂质。然后将各组细胞用RIPA裂解液(Sigma,R0278)在冰上裂解1 h后,在 4 ℃、13 000 r·min-1离心20 min。在RIPA蛋白裂解液中添加混合的蛋白酶抑制剂(Thermo,87786)防止蛋白质降解。蛋白质浓度通过BCA蛋白质测定法测定。通过SDS-PAGE分离等量的蛋白质(30 μg)并半干转到PVDF膜(Millipore,USA)。用5%脱脂奶粉封闭后,分别用抗C3d抗体(CST,97425)、抗c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)JNK抗体(ABcam,ab199380)、抗P-JNK抗体(ABcam,ab278616)和抗GAPDH(ABcam,ab9484)在4 ℃孵育过夜。TBST洗涤3次后,用种属对应的辣根过氧化物酶标记的二抗室温孵育2 h。洗涤后,使用增强型化学发光(ECL)系统(GE Healthcare Life Sciences,USA)可视化蛋白条带。
1.8 统计学方法各组数据以均数±标准差描述,采用GraphPad Prism 8.0软件(GraphPad software,La Jolla,CA)进行统计分析。单因素方差分析(ANOVA)后,两两比较采用Tukey′s post-hoc test,单一比较通过未配对t检验评估。
2 结果
2.1 GSPs对原代星形胶质细胞活力的影响不同浓度的GSPs干预24 h后,分别采用LDH法和MTT法检测了药物对原代AS毒性和细胞增殖的影响。结果表明低、中浓度的(2.5、5、10、20和30 mg ·L-1)GSPs对原代AS没有细胞毒性,也不影响细胞的增殖,但高浓度(35、40和50 mg·L-1)对细胞有明显的细胞毒性(Fig 1A),且抑制细胞的增殖(Fig 1B)。这表明GSPs的给药浓度在30 mg ·L-1之内对原代AS的活力没有影响,为后续实验药物浓度的确定提供了依据。
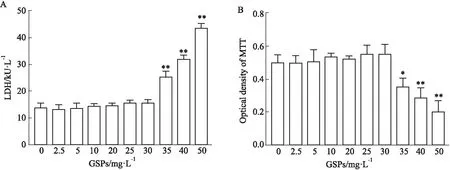
Fig 1 Low concentration of GSPs had no effect on viability of primary AS LDH assay and MTT assay were used to detect the toxicity of different concentrations of GSPs to primary AS. *P<0.05,**P<0.01 vs 0 mg·L-1
2.2 GSPs在体外氧自由基清除实验为了确定GSPs的最优给药浓度,检测了GSPs的抗氧化作用,基于细胞活力实验结果,GSPs的最大给药浓度为35 mg·L-1。与Vc组相比,GSPs在体外对DPPH和ABTS自由基具有强大的清除作用,且具有浓度依赖性,且在30 mg·L-1时清除率分别达到95%和92%(Fig 2)。因此,将GSPs的给药浓度确定为30 mg·L-1。
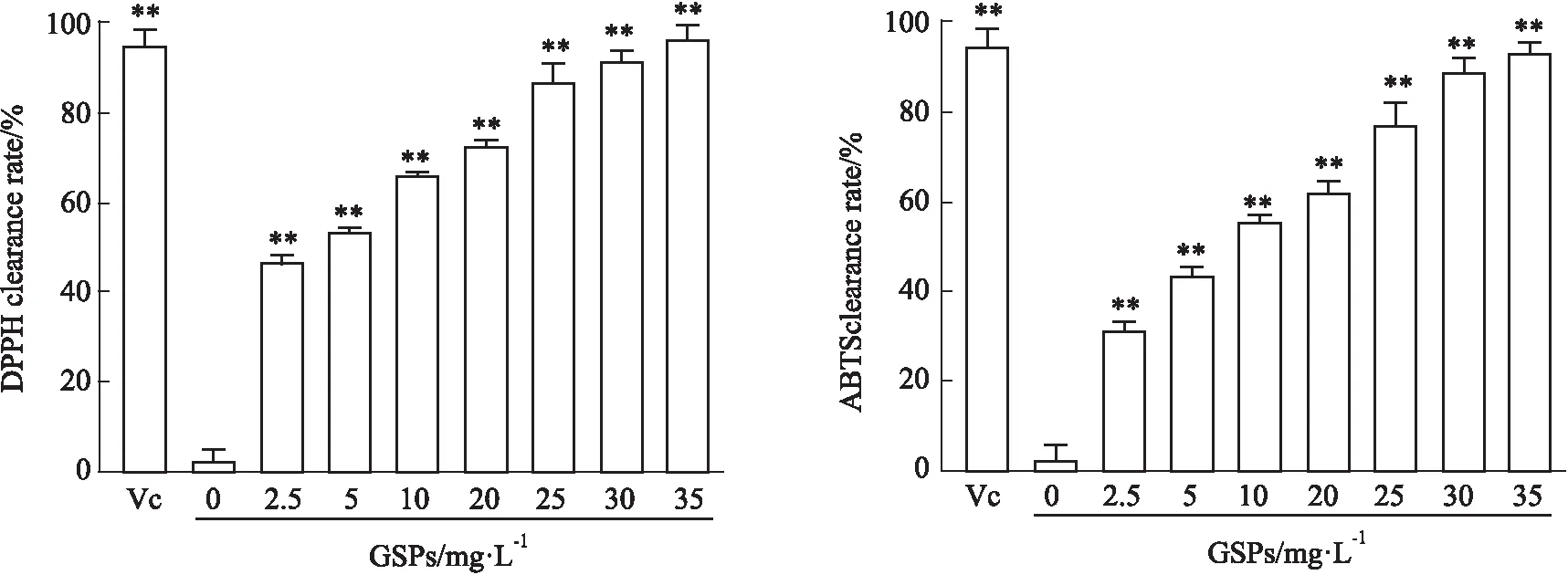
Fig 2 GSPs had a strong scavenging effect on DPPH and ABTS free radicals in vitro in a concentration-dependent manner**P<0.01 vs Vc.
2.3 GSPs对原代AS极化的影响为了研究GSPs对AS极化的影响,从新生小鼠中提取分离原代AS,并使用免疫荧光法测定AS特异性标记物GFAP。如Fig 3A所示,绝大多数细胞呈GFAP阳性,显绿色荧光。DAPI染核,胞核呈蓝色,为总细胞数。用IL-1α、TNF-α和C1q孵育原代AS,发现A1型标记C3d和C1q在基因水平明显增加(Fig 3B)。进一步对蛋白水平进行了检测,发现与正常组相比,模型组C3d和C1q的表达明显升高(Fig 3C、3D),表明A1型AS模型成功建立。为了探索GSPs对AS极化作用,用30 mg·L-1的GSPs干预A1型AS的极化,发现药物可显著降低C3d和C1q的表达(Fig 4A、4B),抑制其向A1型极化。
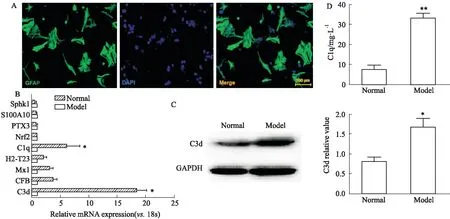
Fig 3 GSPs inhibited AS polarization to type A1
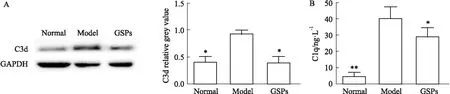
Fig 4 GSPs inhibited AS polarization to type A1
2.4 GSPs对A1型AS分泌的细胞因子的影响研究表明,A1型AS可通过分泌细胞因子发挥作用。因此,我们检测了GSPs(30 mg·L-1)对其分泌的细胞因子的影响。发现GSPs明显抑制促炎因子TNF-α、IL-1α和IL-6的分泌(Fig 5A),减少氧化应激因子H2O2的表达(Fig 5B),促进抑炎因子TGF-β和神经营养因子CNTF(Fig 5C)的分泌。
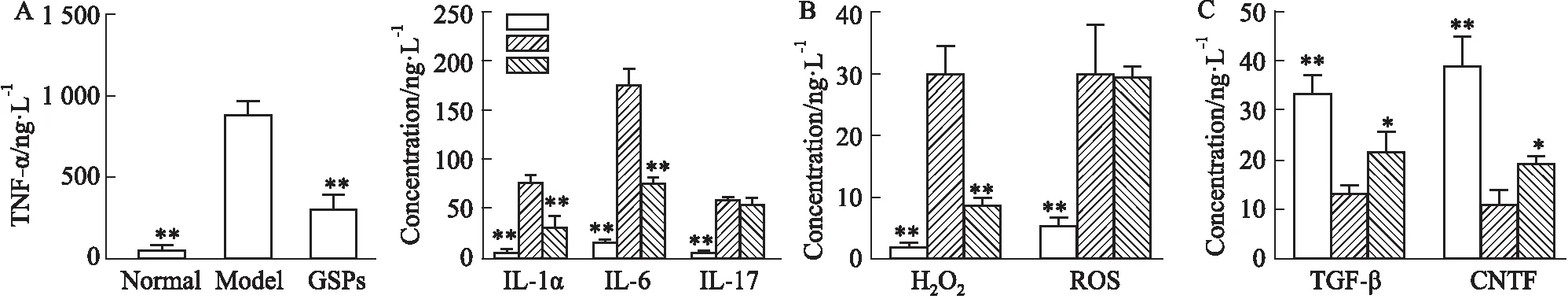
Fig 5 GSPs inhibited secretion of inflammatory factors and promoted secretion of anti-inflammatory factors and neurotrophic factors
2.5 GSPs对A1型星形胶质细胞JNK的影响研究表明,IL-1α、TNF-α和C1q激活的AS中JNK信号通路被激活,与AS的炎症反应有关,因此,接下来检测了GSPs对JNK的影响,发现GSPs明显下调了AS中JNK的磷酸化水平(Fig 6)。
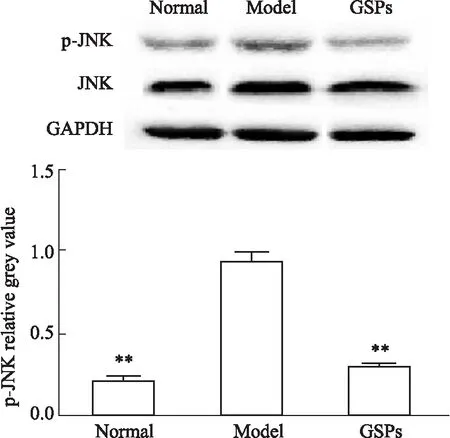
Fig 6 Phosphorylation of JNK in AS inhibited by GSPs was determined by Western blot**P<0.01 vs Model.
3 讨论
神经炎性脱髓鞘疾病中,大量的反应性AS增殖,虽然其作用机制尚不清楚,但Barres等[13]根据其表型和功能的变化,发现神经炎症和缺血诱导了两种不同类型的反应性AS,并将其命名为A1型和A2型。A1型AS大量存在于阿尔茨海默病、帕金森病、肌萎缩侧索硬化症和MS中[3,14],并丧失了促进神经元存活、突触重塑和吞噬的能力,同时分泌诸多神经毒性因子损伤神经元和髓鞘结构。如在EAE模型中,A1型可以通过高表达乳糖神经酰胺,促进髓鞘脱失和加重中枢炎症的效应[5]。与之相反,A2型则上调了许多神经营养因子的表达,促进CNS受损组织的恢复和修复。如A2型可分泌CXCL8、CXCL1和CXCL10等趋化因子募集OPCs至脱髓鞘部位、高表达基质金属蛋白酶-1(metalloproteinase-1,TIMP-1)和脑源性神经营养因子(brain-derived neurotrophic factor,BDNF)促进髓鞘再生[13,15]。因此,抑制A1型或诱导A2型反应性AS可能对疾病产生有益的影响。本研究采用促炎因子C1q、TNF-α和IL-1α诱导高表达C3d的A1型AS,而GSPs可明显抑制C3d的表达,表明GSPs可能通过抑制A1型AS缓解髓鞘脱失。
研究发现,属于丝裂原活化蛋白激酶(mitogen-activated protein kinase,MAPK)的JNK信号通路,可以参与AS的激活[16]。我们的实验证明了C1q、TNF-α和IL-1α诱导的A1型AS中存在JNK磷酸化的明显增加,提示JNK信号通路的活化可能与AS的极化有关。而GSPs是原花青素低聚体的混合物,包括儿茶素、表儿茶素和原花青素B2等有效成分,采用网络药理学的方法预测其作用靶点,发现GSPs抑制中枢神经氧化应激和炎性因子的机制可能与JNK的磷酸化有关[17]。本研究发现GSPs能够降低A1型AS中JNK的磷酸化,抑制AS向A1型极化,说明减少JNK的磷酸化GSPs影响AS极化的靶点,为进一步研究GSPs的作用机制奠定了基础。
反应性AS的表型是动态变化的,其极化趋势主要取决于微环境的不同刺激。如Barres等报道异常活化的MG可通过分泌C1q、TNF-α和IL-1α作用于AS,并诱导其向A1型极化。A1型AS通过分泌IL-1α、TNF-α、IL-6、IL-17和C1q等促炎因子及H2O2和ROS等氧化应激因子攻击和损伤神经组织的同时,还可以反作用于A1型AS加重和促进继发性炎症反应。如IL-17与其受体结合,可激活AS下游的核因子(nuclear factor κB,NF-κB),导致促炎因子的产生。而抗炎因子如IL-4、IL-10和TGF-β等,可以逆转A1型AS,激活AS的神经保护作用,诱导其向A2型极化[18]。如TGF-β信号能介导NF-κB的抑制,从而缓解中风后的神经炎症。因此,有目的性地改变反应性AS的微环境,有利于获得具有神经保护作用的表型。为了研究GSPs抑制A1型AS极化的作用机制,我们进一步检测了体系中相关的炎性因子、氧化应激因子、抗炎因子和神经营养因子。结果表明,GSPs可以明显减少IL-1α、TNF-α、IL-6、IL-17和C1q的分泌,抑制H2O2的表达,同时提高TGF-β和CNTF的水平,说明GSPs抑制AS向A1型极化与其能改善促炎微环境密切相关。但本次实验仅在体外探索了GSPs对AS的直接作用,由于在机体内AS的极化是由多种因素共同决定的,所以我们将进一步研究GSPs是否可以通过影响其它细胞产生效应。
综上所述,本研究发现GSPs可明显抑制AS向A1型极化,作用机制与其显著抑制JNK的磷酸化有关。此外,GSPs还能减少炎性因子和氢自由基的产生,促进抗炎因子和神经营养因子的分泌,进而改善炎症微环境,发挥神经保护作用。
猜你喜欢
中华耳科学杂志(2022年1期)2022-11-24
磁共振成像(2022年8期)2022-10-08
现代财经-天津财经大学学报(2022年5期)2022-06-01
航天电子对抗(2022年2期)2022-05-24
北京航空航天大学学报(2021年9期)2021-11-02
航天电子对抗(2019年4期)2019-06-02
党的生活(黑龙江)(2018年9期)2018-10-17
中成药(2018年9期)2018-10-09
益寿宝典(2018年1期)2018-01-27
中国实验动物学报(2017年2期)2017-05-18
