
利拉鲁肽通过调控自噬和Na+,K+-ATPase活性抑制高糖诱导的心肌细胞肥大
2023-01-05 08:11野战鹰杨林泉马慧娟
中国药理学通报 2023年1期
张 哲,野战鹰,王 杏,杨林泉,马慧娟
(河北省人民医院 1. 代谢病重点实验室、2. 神经外三科,河北 石家庄 050051)
近年来,糖尿病患病率持续增高,预计截至2030年全球范围内糖尿病患病人数将达到3亿,而心血管并发症是糖尿病患者致死和致残的主要原因。心室重构是糖尿病心肌病(diabetic cardiomyopathy,DCM)的重要病理机制,而心肌肥厚是心室重构的起始阶段,持续的心肌肥厚进一步加重心脏收缩和/或舒张功能障碍,最终发展为心力衰竭[1]。因此,探索逆转心肌肥厚的新靶点对于延缓DCM进展,降低心力衰竭致死率具有重要临床意义。
自噬是真核生物细胞内的一种自我保护机制,可通过溶酶体降解途径清除受损的细胞器、错误折叠的蛋白、应激产物等,在维持细胞稳态、促进能量再生方面发挥重要作用。自噬障碍与代谢性疾病发病密切相关,如肥胖、胰岛素抵抗、糖尿病、动脉粥样硬化等。近来研究发现,在多种心肌肥厚模型中均存在自噬缺陷,而增强AMPK/mTOR通路介导的自噬可逆转心肌肥厚,提示调控自噬活性有可能成为减轻心肌肥厚的潜在策略[2-3]。
Na+,K+-ATPase(NKA)是哺乳动物细胞膜上普遍存在的一种跨膜蛋白,在维持离子平衡、静息电位及肌细胞兴奋性方面发挥重要调控作用。已证实NKA活性下降可引起细胞内Na+、Ca2+积聚,从而促进肥厚相关基因的表达,导致心肌肥厚发生,而上调NKA活性能够抑制心肌肥厚[4]。新近研究表明,自噬与NKA之间存在相关性,激活自噬可通过调控NKA活性影响疾病发生发展。Singh等[5]报道,自噬诱导剂雷帕霉素可减轻氧化应激损伤并上调NKA活性,从而延缓神经退行性变进程。Liu等[6]也提示,激活自噬可减轻缺血/再灌注所致肾损伤,并显著提高NKA等Na+转运体活性;利用自噬抑制剂3-甲基腺嘌呤(3-MA)或敲除自噬标志基因Atg5能够阻断这一作用。但是,自噬能否通过调控NKA抑制糖尿病心肌肥厚仍未可知。
利拉鲁肽(liraglutide,LRG)是一种新型降糖药,可通过抗炎、抗氧化、抗凋亡等机制发挥心血管保护作用。本课题组前期研究表明,LRG可调控Sirt1/AMPK通路减轻DCM大鼠心肌损伤[7]。本研究建立高糖诱导的H9c2心肌细胞肥大模型,探讨LRG对心肌细胞肥大的保护作用及其与自噬、NKA间的关系,并进一步阐明其分子机制。
1 材料与方法
1.1 药物与试剂大鼠心肌H9c2细胞株,购自中国科学院典型培养物保藏委员会细胞库。LRG(国药准字J20160037),诺和诺德;罗丹明标记鬼笔环肽(CA1610)、单丹磺胺戊二胺(MDC)试剂盒(G0170),索莱宝;FastKing cDNA第一链合成试剂盒(U8918),天根;Na+,K+-ATPase(NKA)测定试剂盒(A070-2-2),建成;ANP(sc-515701),Santa Cruz;β-MHC(ab207926)、Beclin-1(ab62557)、NKA α1(ab7671)、NKA α2(ab166888),Abcam;LC3(CST4108)、p62(CST5114)、Sirt1(CST8469)、AMPK(CST2532)、p-AMPK(CST2535)、mTOR(CST5536)、p-mTOR(CST2983),Cell Signaling Technology;3-MA(T1879),Targetmol;EX 527(S1541)、Compound C(CC,S7840),Selleck。
1.2 仪器HERA cell-150i二氧化碳培养箱、ND-2000紫外分光光度计,Thermo Fisher Scientific;AXOVERT25C倒置显微镜,Zeiss;DMI3000B荧光显微镜,Leica;Fresco17冷冻高速离心机,Thermo;7500实时荧光定量PCR仪,ABI;JY300HE电泳仪,君意;MiniChemi 610 Plus凝胶成像系统,赛智。
1.3 细胞培养及分组H9c2细胞在含10%胎牛血清的DMEM-F12培养基中,于37 ℃、5% CO2培养箱中培养,待细胞达80%融合时收集细胞进行实验。细胞分为对照(CON)组(5.5 mmol·L-1葡萄糖处理细胞48 h)、高糖(HG)组(33.3 mmol·L-1葡萄糖处理细胞48 h)、低、中、高剂量LRG(LRG-L、LRG-M、LRG-H)组(分别用25、50、100 nmol·L-1LRG和33.3 mmol·L-1葡萄糖共同处理细胞48 h)、LRG-H+自噬抑制剂3-MA组(1 mmol·L-13-MA作用细胞24 h后,用100 nmol·L-1LRG和33.3 mmol·L-1葡萄糖共同处理细胞48 h)、LRG-H+Sirt1抑制剂EX 527组(200 nmol·L-1EX 527作用细胞24 h后,用100 nmol·L-1LRG和33.3 mmol·L-1葡萄糖共同处理细胞48 h)、LRG-H+AMPK抑制剂CC组(20 μmol·L-1CC作用细胞24 h后,用100 nmol·L-1LRG和33.3 mmol·L-1葡萄糖共同处理细胞48 h)。
1.4 鬼笔环肽染色细胞接种于24孔板,待细胞生长至50%时,4%多聚甲醛室温固定10 min,PBS清洗2次,0.5% Triton X-100透化处理5 min,PBS清洗2次,加入终浓度为200 nmol·L-1的鬼笔环肽染料,室温避光孵育30 min,DAPI染核5 min,荧光显微镜观察心肌细胞骨架微丝,拍照。每组随机选取50个细胞,ImageJ测定细胞表面积。
1.5 NKA活性测定细胞接种于6孔板,待细胞生长至80%~90%时,收集各组细胞至离心管中,制备细胞匀浆,20 000×g离心30 min,弃上清液,分离肌膜成分,按照试剂盒说明书检测细胞膜NKA活性。
1.6 MDC染色细胞接种于24孔板,待细胞生长至50%时,弃去培养基,Wash buffer清洗2次,每孔加入MDC染液100 μL,室温避光孵育45 min,弃去染液,Wash buffer清洗2次,荧光显微镜观察自噬囊泡,计数并拍照,计算阳性细胞比例。
1.7 Real time PCRTRIzol提取心肌细胞总RNA,用FastKing cDNA第一链合成试剂盒合成cDNA。NKA α1引物,F:5′-GCTGTCGTCATCATAACTGGC-3′,R:5′-GCTCATCTTCTCTCCATTTCG-3′;NKA α2引物,F:5′-TCATCGGCATCATCGTAGC-3′,R:5′-CGTCAGGCACACAGTAACAGT-3′;GAPDH引物,F:5′-TGAACGGGAAGCTCACTG-3′,R:5′-GCTTCACCACCTTCTTGATG-3′。在实时荧光定量PCR仪上进行扩增反应,2-ΔΔCt法分析NKA α1、NKA α2 mRNA相对表达量。
1.8 Western blot提取各组细胞总蛋白,BCA法定量后进行SDS-PAGE电泳,将目的蛋白转至PVDF膜,5%脱脂奶粉封闭2 h,加入特异性一抗(ANP 1 ∶600,β-MHC 1 ∶600,NKA α1 1 ∶1 000,NKA α2 1 ∶1 000,Beclin-1 1 ∶500,LC3 1 ∶1 000,p62 1 ∶1 000,Sirt1 1 ∶1 000,AMPK 1 ∶1 000,p-AMPK 1 ∶1 000,mTOR 1 ∶1 000,p-mTOR 1 ∶1 000),4 ℃冰箱孵育过夜,洗膜后加入辣根过氧化物酶标记的IgG(1 ∶5 000稀释),室温孵育1 h,ECL显影剂显色后用ImageJ进行灰度分析。
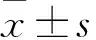
2 结果
2.1 LRG对高糖诱导的心肌细胞肥大的影响鬼笔环肽染色显示(Fig 1A):与CON组相比,HG组细胞表面积明显增加(P<0.01),不同剂量LRG干预后细胞表面积较HG组明显降低(P<0.01),而自噬抑制剂3-MA可部分拮抗LRG的作用(P<0.01)。Western blot显示(Fig 1B):与CON组相比,HG组ANP、β-MHC蛋白表达明显增加(P<0.01),不同剂量LRG干预后ANP、β-MHC蛋白表达较HG组明显降低(P<0.01),而自噬抑制剂3-MA可部分拮抗LRG的作用(P<0.01)。
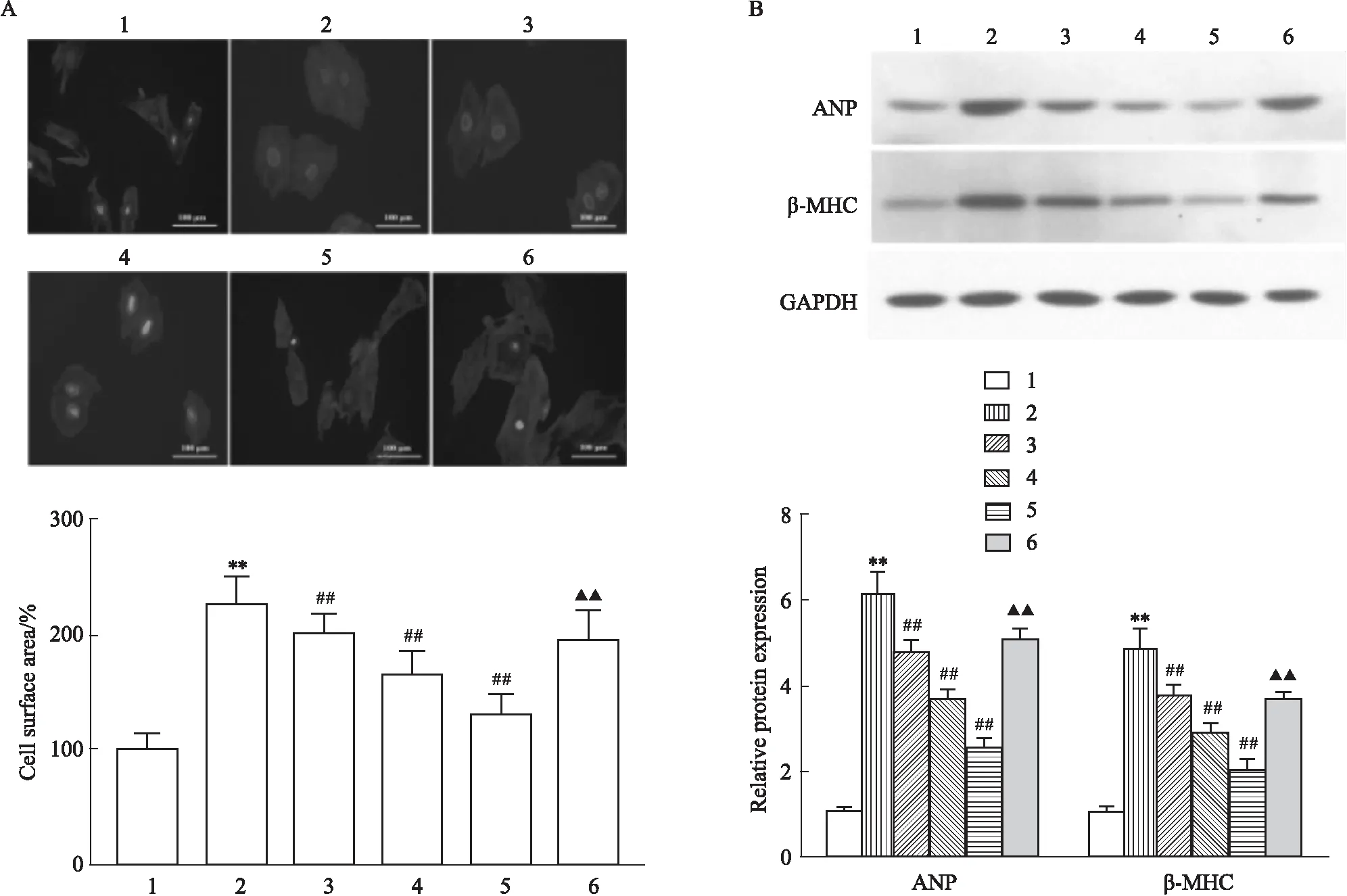
Fig 1 Effects of LRG on HG-induced cardiomyocyte hypertrophy
2.2 LRG对高糖诱导的心肌细胞NKA活性和NKA α亚基表达的影响NKA活性测定显示(Fig 2A):与CON组相比,HG组NKA活性明显降低(P<0.01),不同剂量LRG干预后NKA活性较HG组明显增加(P<0.05或P<0.01),而自噬抑制剂3-MA可部分拮抗LRG的作用(P<0.01)。文献报道,NKA最小功能单位为αβ异源二聚体,其中α亚基是NKA的催化亚基,包括α1~α4四种亚型,啮齿类动物心肌仅表达NKA α1、α2,本研究进一步检测NKA α1、α2表达情况。Real time PCR和Western blot显示(Fig 2B-C):各组NKA α2 mRNA和蛋白表达变化趋势与NKA活性测定结果一致,而NKA α1 mRNA和蛋白表达相比无统计学差异(P>0.05)。
2.3 LRG对高糖诱导的心肌细胞自噬的影响MDC染色显示(Fig 3A):与CON组相比,HG组自噬囊泡数量明显降低(P<0.01),不同剂量LRG干预后自噬囊泡数量较HG组明显增加(P<0.01),而自噬抑制剂3-MA可部分拮抗LRG的作用(P<0.01)。Western blot显示(Fig 3B):与CON组相比,HG组Beclin-1、LC3-Ⅱ/LC3-Ⅰ蛋白表达明显降低,p62蛋白表达明显增加(P<0.01),不同剂量LRG干预后,Beclin-1、LC3-Ⅱ/LC3-Ⅰ蛋白表达较HG组明显增加,p62蛋白表达较HG组明显降低(P<0.05或P<0.01),而自噬抑制剂3-MA可部分拮抗LRG的作用(P<0.01)。
2.4 阻断Sirt1/AMPK/mTOR信号通路逆转LRG对高糖诱导的心肌细胞肥大的抑制作用鬼笔环肽染色显示(Fig 4A):与CON组相比,HG组细胞表面积明显增加(P<0.01),高剂量LRG干预后细胞表面积较HG组明显降低(P<0.01),而Sirt1抑制剂EX 527、AMPK抑制剂CC可部分拮抗LRG的作用(P<0.01)。同时,Western blot也证实(Fig 4B):Sirt1抑制剂EX 527、AMPK抑制剂CC可减弱高剂量LRG下调ANP、β-MHC蛋白表达的作用(P<0.01)。
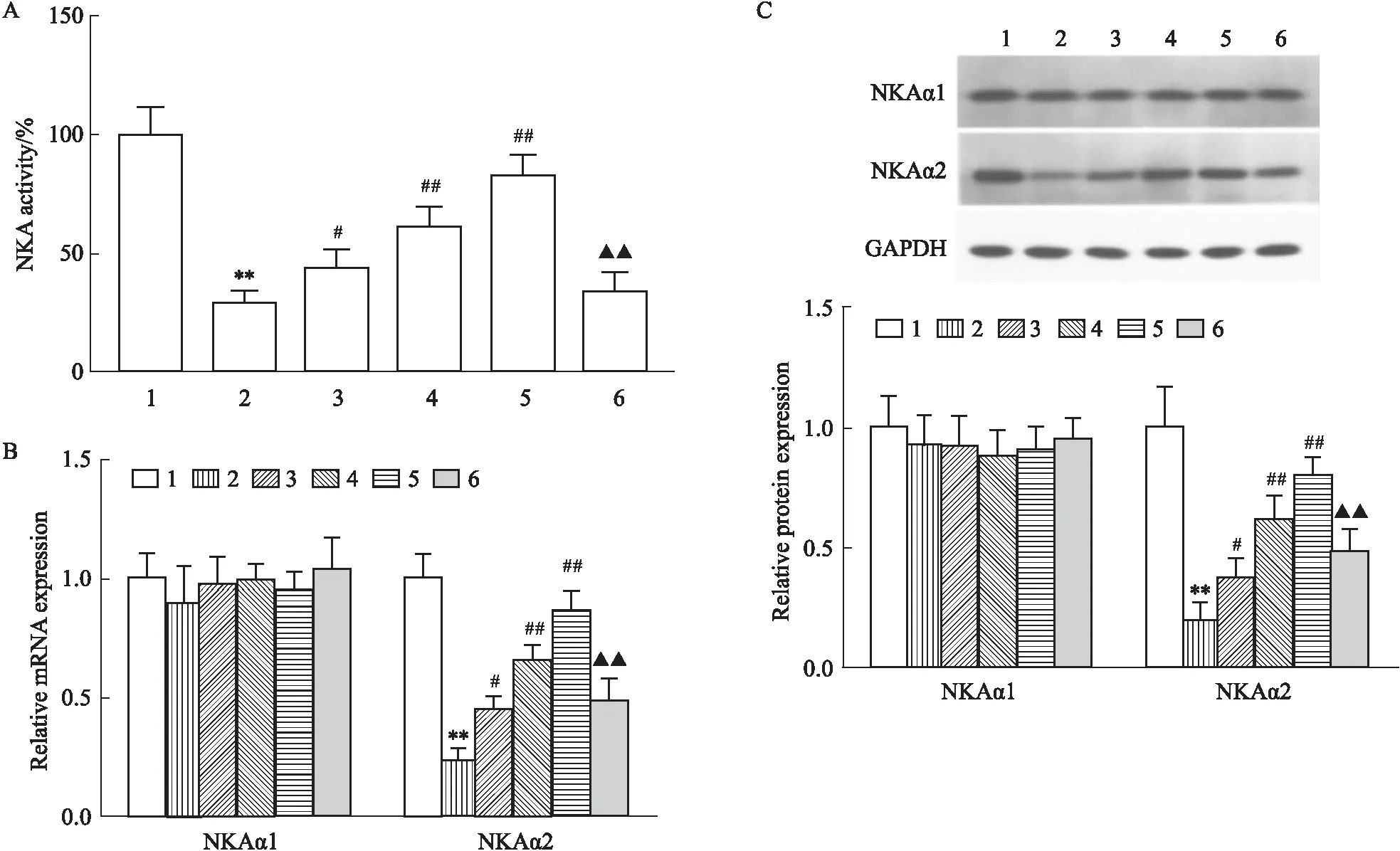
Fig 2 Effects of LRG on NKA activity and NKA αexpression in HG-induced H9c2 cells
Fig 3 Effects of LRG on autophagy in HG-induced H9c2 cells

Fig 4 Inhibition of Sirt1 or AMPK reversed LRG-mediated cardioprotective effect in HG-exposed H9c2 cells
2.5 阻断Sirt1/AMPK/mTOR信号通路逆转LRG对高糖诱导的心肌细胞NKA活性和NKA α2亚基表达的上调作用NKA活性测定显示(Fig 5A):与CON组相比,HG组NKA活性明显降低(P<0.01),高剂量LRG干预后NKA活性较HG组明显增加(P<0.01),而Sirt1抑制剂EX 527、AMPK抑制剂CC可部分拮抗LRG的作用(P<0.01)。同时,Real time PCR和Western blot也证实(Fig 5B-C):Sirt1抑制剂EX 527、AMPK抑制剂CC可减弱高剂量LRG上调NKA α2 mRNA和蛋白表达的作用(P<0.01)。

Fig 5 Inhibition of Sirt1 or AMPK reversed LRG-mediated increase of NKA activity and up-regulation of NKA α2 in HG-exposed H9c2 cells
2.6 阻断Sirt1/AMPK/mTOR信号通路逆转LRG对高糖诱导的心肌细胞自噬的上调作用MDC染色显示(Fig 6A):与CON组相比,HG组自噬囊泡数量明显降低(P<0.01),高剂量LRG干预后自噬囊泡数量较HG组明显增加(P<0.01),而Sirt1抑制剂EX 527、AMPK抑制剂CC可部分拮抗LRG的作用(P<0.01)。同时Western blot也证实(Fig 6B):Sirt1抑制剂EX 527、AMPK抑制剂CC可减弱高剂量LRG上调Sirt1、p-AMPK/AMPK、Beclin-1蛋白表达及下调p-mTOR/mTOR蛋白表达的作用(P<0.01)。
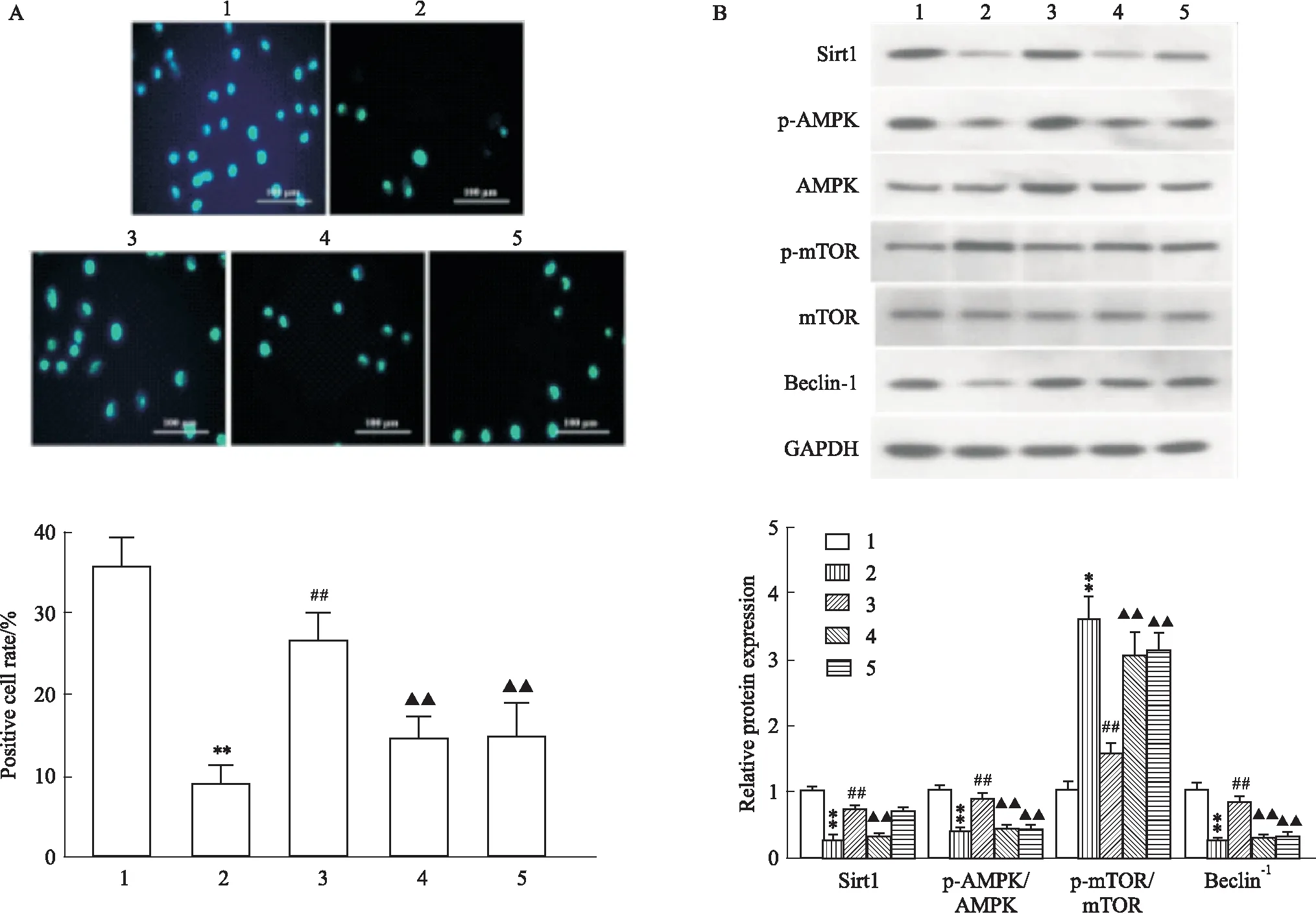
Fig 6 Inhibition of Sirt1 or AMPK reversed LRG-mediated activation of Sirt1/AMPK/mTOR signaling and autophagy in HG-exposed H9c2 cells
3 讨论
2型糖尿病是以血糖异常升高为特征的慢性代谢性疾病,持续的高血糖被认为是导致DCM、糖尿病肾病、糖尿病视网膜病变等慢性并发症的独立危险因素。LRG是一种新型胰高血糖素样肽1类似物,因其良好的降糖效果被广泛应用于2型糖尿病的临床治疗中,且对多种心血管疾病具有显著保护作用,如高血压、动脉粥样硬化、心力衰竭等。但LRG对糖尿病心肌肥厚的作用及机制报道尚少。既往研究发现,高浓度葡萄糖刺激下H9c2心肌细胞,具有与原代培养的胎鼠心肌细胞相似的肥厚性反应,表现为肥大标志基因(ANP、β-MHC、CTGF等)mRNA表达快速上调。本研究中,我们采用33.3 mmol·L-1葡萄糖作用于H9c2细胞48 h,观察到细胞表面积显著增加,同时ANP、β-MHC蛋白表达显著上调,证实糖尿病心肌肥厚模型构建成功;LRG干预能够抑制心肌细胞肥大,提示LRG可能成为治疗该病的有效药物。
大量研究显示,NKA是细胞排出Na+的唯一途径,其活性降低造成细胞内Na+积聚,继而通过促进Na+/Ca2+交换体(NCX)反向转运模式导致细胞内Ca2+浓度增加。持续升高的细胞内Ca2+是诱导心肌细胞肥厚性生长的重要病理机制,并导致心功能障碍和心力衰竭进一步加重[8]。本研究表明,高糖处理后心肌细胞NKA活性、NKA α2 mRNA和蛋白表达降低;给予LRG干预后,NKA活性、NKA α2 mRNA和蛋白表达得以升高。可见,LRG对糖尿病心肌肥厚的治疗机制与促进NKA活性恢复及上调NKA α2表达有关。同时,Real time-PCR和Western blot检测到NKA α1 mRNA和蛋白表达在各组间均无统计学差异,说明糖尿病心肌肥厚发生发展并不涉及NKA α1变化。我们分析NKA α1、α2在心肌肥厚中的不同作用与其在心肌细胞膜的分布不同有关。多项研究表明,α1广泛表达在表面肌膜,可将细胞内Na+维持在整体较低的水平,在调控离子稳态方面发挥“管家作用”;而α2与NCX、L型Ca2+通道均集中表达在横小管,且α2对其内源性抑制剂强心苷的亲和力较α1高4倍,因此局部Na+水平升高即可通过调控毗邻的NCX和内质网Ca2+通道引起Ca2+的大量释放,可见α2在调控心肌收缩力方面发挥关键作用[9-10]。已证实NKA α2表达下调是异丙肾上腺素导致的心肌细胞NKA活性降低和心肌肥厚的重要因素[11]。在心肌梗死小鼠模型中,特异性过表达NKA α2的转基因小鼠心肌细胞Ca2+瞬变幅度较WT小鼠显著降低,可逆转心室重构,并阻止心功能恶化[12]。因此,LRG可能通过上调NKA α2表达增强NKA活性,从而逆转高糖诱导的心肌肥厚。
目前,自噬在心肌肥厚中的作用仍存在争议。有学者认为,增强自噬活性可能通过增加ATP合成、降解错误折叠的蛋白逆转心肌肥厚,延缓心力衰竭进程。然而,过度自噬可能促进细胞凋亡加重心室重构,采用自噬抑制剂3-MA或siRNA干扰技术下调自噬相关基因表达后,心肌肥厚得以明显减轻[13]。本研究结果显示,高糖诱导心肌细胞自噬缺陷,LRG干预通过调控自噬相关基因表达增强自噬水平,提示LRG的心脏保护作用可能是通过调控自噬实现的。我们推测,自噬在心肌肥厚中发挥保护性作用还是有害作用与心室重构的不同阶段、造模方法及涉及的信号通路有关。
Sirt1/AMPK通路在调控炎症、凋亡、自噬、能量代谢及心脏保护性蛋白表达方面发挥重要作用。Li等[14]研究发现,Sirt1/AMPK通路活化后能够发挥抗凋亡、抗氧化作用,减轻血管紧张素II(Ang II)诱导的心肌肥厚和心功能障碍。值得注意的是,过表达Sirt1或应用AMPK激动剂AICAR可逆转晚期糖基化终产物诱导的心肌细胞NKA活性降低,提示Sirt1/AMPK通路可调控NKA活性[15]。本研究发现,给予特异性Sirt1抑制剂EX 527、AMPK抑制剂CC后,LRG对NKA活性、NKA α2 mRNA和蛋白表达的上调作用及心肌肥大的保护作用被阻断了,同时伴有自噬囊泡数量减少及自噬标志基因Beclin-1表达下调,提示LRG可能通过激活Sirt1/AMPK/mTOR自噬信号促进NKA活性恢复,从而逆转高糖诱导的心肌细胞肥大。然而,关于心肌细胞中自噬信号通路调控NKA分子机制的研究尚少。目前已有研究指出NKA和AMPK之间存在直接联系。Xiong等[16]研究发现,靶向NKA胞外结构域的抗体DR-Ab通过上调NKA活性减轻Ang II诱导的心肌肥厚,该细胞保护作用涉及AMPK信号通路的活化。Pirkmajer等[17-18]在骨骼肌细胞、肾小管细胞中也证实,AMPK的持续活化促使NKA去磷酸化,从而抑制NKA的内吞并保护其免遭溶酶体降解。此外,糖尿病状态下线粒体产生的自由基(ROS)可引起NKA结构改变和氧化损伤,从而影响线粒体氧化磷酸化并降低细胞内ATP的合成,这是导致NKA活性降低的重要因素。增强自噬活性能够直接清除功能异常的线粒体和ROS,同时使细胞内ATP含量增加,从而促进NKA活性恢复。以上研究提示,自噬信号通路可能通过多种途径调控NKA。
综上,本研究表明,LRG能够抑制高糖诱导的心肌细胞肥大,其机制可能与调控Sirt1/AMPK/mTOR自噬信号增强NKA活性有关。本研究证实了LRG的体外抗肥大作用,也为将自噬信号介导的NKA调控作为防治DCM的分子靶点提供了实验依据。不足之处在于,本研究尚未开展在体实验进一步验证此结论。今后我们将利用DCM动物模型进行相关研究,旨在探索和完善LRG的心脏保护作用及机制。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年7期)2021-11-04
天然产物研究与开发(2018年7期)2018-08-21
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
中成药(2017年8期)2017-11-22
上海农业学报(2017年3期)2017-04-10
中成药(2016年8期)2016-05-17
中国病理生理杂志(2015年8期)2015-12-21
中国当代医药(2015年16期)2015-03-01
