
人尿激肽原酶对SAMP8小鼠模型认知功能的影响及机制
2023-01-05 08:11:24王智亮高世超张跃其
中国药理学通报 2023年1期
王智亮,赵 莉,李 靖,高世超,张跃其
(潍坊市人民医院 1.药物临床研究中心、2.疼痛科,山东 潍坊 261000;3.纽约州立大学布法罗分校药理系,纽约州 布法罗 14214;4.潍坊市人民医院神经内科,山东 潍坊 261000)
阿尔茨海默病(Alzheimer′s disease,AD)是一种神经退行性疾病,是老年人群中导致痴呆的主要原因。超过95%的AD患者是散发性AD(sporadic AD,SAD)[1]。随着我国医疗条件的改善,老年人口比例的上升,SAD患者逐年增多。目前AD尚无有效的治疗方法,因此,针对SAD的治疗研究日显重要和紧迫。针对SAD的药物治疗研究中,动物模型是药物筛选的重要工具,SAMP8小鼠是目前公认的非转基因痴呆模型[2]。本课题组前期发现,8月龄SAMP8小鼠海马神经炎性斑、细胞外β淀粉样蛋白42(Aβ42)、p181-Tau增多,认知功能明显下降,证实SAMP8小鼠可作为SAD理想的动物模型[3]。
人尿激肽原酶(human urinary kallidinogenase,HUK)是激肽释放酶—激肽系统的调节物质,能选择性扩张脑细小动脉,减轻炎性反应及氧化应激水平,促进小血管生成,并减少神经元及神经胶质细胞凋亡[4]。已有研究报道,HUK可改善大鼠脑缺血再灌注后空间学习记忆能力[5],而记忆能力下降与突触可塑性密切相关,但目前尚无关于HUK对SAD中认知能力影响及突触相关蛋白的研究报道。本研究通过观察SAMP8小鼠给予HUK干预后的行为学及脑组织蛋白改变,探讨HUK对SAD模型认知功能的影响及机制。
1 材料与方法
1.1 实验材料8月龄♂SAMP8小鼠和SAMR1小鼠购自山东大学实验动物中心[SCXK(鲁) 2019 - 0001],动物实验遵循动物实验伦理要求。HUK购自广东天普生化医药股份有限公司(0.15PNAU/瓶,粉针剂)。胆碱乙酰转移酶(choline acetyltransterase,ChAT)购自Mlillipore公司,稀释度为1 ∶100,二抗为羊抗免IgG,DAB显色试剂盒(北京中杉金桥公司)。IL-1β、IL-18 ELISA试剂盒购自江苏碧云天生物科技有限公司;MPO检测盒购自南京建成生物工程研究所,引物由上海生工公司构建。RNA提取试剂、逆转录试剂盒、RtPCR扩增试剂盒购自上海吉凯基因技术有限公司。PSD95、BDNF抗体购自Abcam公司。SYN、pCREB抗体购自Cell Signaling公司。Morris水迷宫购自北京大学医学部神科所。冰冻切片机购自德国Leica公司。
1.2 试验方法
1.2.1动物分组及给药 未处理的SAMR1小鼠作为对照组(SAMR1组)。SAMP8鼠随机分为5组:SAMP8组、治疗组(分别给予8.75×10-3、1.75×10-2、3.5×10-2、7.0×10-2PNAU·kg-1尿激肽原酶,对应a、b、c、d组),每组10只,每日同一时间(9 ∶00 am)舌下静脉注射0.9%氯化钠溶液稀释的HUK,氯化钠量以1 mL·kg-1计。SAMR1组和SMAP8组均给予0.9%氯化钠溶液1 mL·kg-1,1次/日,给药共7 d,给药结束后进行行为学实验。
1.2.2Morris水迷宫实验 每日游泳5次,每次60 s,记录小鼠逃避潜伏期。若小鼠在60 s内未找到平台,则引导至目标平台停留5 s,观察周围环境。实验历时6 d。d 7撤掉平台,选择象限I为入水点,水中游泳60 s,记录动物在目标象限(target quadrant)的游泳时间、穿越平台次数(passing times)。
1.2.3组织标本制备 水迷宫结束后各组小鼠进行腹腔注射麻醉,暴露心脏后向左心室内注射1%肝素钠0.2 mL,经左心室行主动脉插管,剪开右心耳后以生理盐水冲洗血管,再以含4%多聚甲醛的0.01 mol·L-1PBS(4 ℃,pH 7.40)250 mL灌注固定,之后取材。留取一侧脑组织行PCR检测,另一侧脑组织放于多聚甲醛中固定,经脱水、石蜡包埋、切片后,进行免疫组化显色。
1.2.4免疫组化检测 免疫组化测定海马组织CA3区ChAT表达。取海马组织切片,脱蜡水化,室温孵育10 min,PBS冲洗后以1% BSA+0.4% PBST封闭1 h,加入兔源多克隆抗ChAT抗体,37 ℃孵育1 h。PBS冲洗后加入羊抗兔的二抗,室温孵育1 h;PBS冲洗后滴加辣根酶标记生物素,孵育20 min后PBS冲洗,行DAB增强显色,梯度酒精脱水,二甲苯透明,中性树胶封片。荧光显微镜下观察CA3区视野。
1.2.5RT-PCR检测 提取总RNA,逆转录cDNA。使用ChAT基因引物进行逆转录。引物序列:上游:5′-GGTGGCCCAGAAGAGCAGTATC-3′,下游:5′-ATTGGAGGCAGGCGTTCATC-3′。PCR循环条件为:94 ℃预变性3 m,94 ℃变性30 s,58 ℃退火30s,72 ℃延伸1 m后进行36个循环,进行融解曲线分析引物的特异性。相对定量2-△△Ct法比较各组间表达差异。
1.2.6MPO活性及IL-1β、IL-18含量测定 各组中任取5只大鼠,取脑组织后剥离海马,以生理盐水制成10%的组织匀浆,取0.5 mL的组织匀浆,按照试剂说明书,采用分光光度计比色法用于测定髓过氧化物酶(MPO)活性,以每克脑组织所含酶活力单位(U)表示(U·g-1)。另取0.5 mL脑组织匀浆,4 ℃离心10 min后取上清液,按照说明书步骤分别用IL-1β、IL-18试剂盒检测组织IL-1β、IL-18含量。
1.2.7Western blot检测 突触相关的学习记忆蛋白表达脑组织中加入裂解液(1 mL·mg-1),匀浆后离心(4 ℃下,10 min)。加入2×缓冲液后煮沸5 min。每孔上样100 μg后进行电泳(30 mA)分离蛋白质后以PVDF膜转膜40 min,使用5%脱脂牛奶进行封闭,室温匀摇2 h,将膜于PSD95一抗(1 ∶600)、SYN一抗(1 ∶800)、BDNF(1 ∶1 000)、pCREB(1 ∶500)于4 ℃孵育过夜,HRP标记二抗孵育1 h,经漂洗后加入ECL发光剂。化学发光分析仪拍照后用ImageJ软件进行半定量分析。
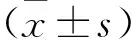
2 结果
2.1 各组小鼠空间认知功能进行水迷宫测试前,各组间小鼠体质量差异无显著性(P>0.05)。进行空间探索实验后,相较于SAMR1组,SAMP8组小鼠的平台穿梭次数减少(P<0.05),在的目的象限内探索时间短(P<0.05),游泳轨迹紊乱盲目,多采为边缘及随机轨迹。而给予8.75×10-3、1.75×10-2、3.5×10-2、7.0×10-2PNAU·kg-1不同剂量HUK后,平台穿梭次数较SAMP8组增多(P<0.05或P<0.01),在目的象限内探索时间短(P<0.05或P<0.01),游泳轨迹目的性强(Fig 1 A~C)。以上结果说明,给予HUK处理后,SAMP8小鼠空间探索能力得到明显提高,空间记忆能力改善。我们以7.0×10-2PNAU·kg-1剂量的d组(HUK组)进行后续实验。
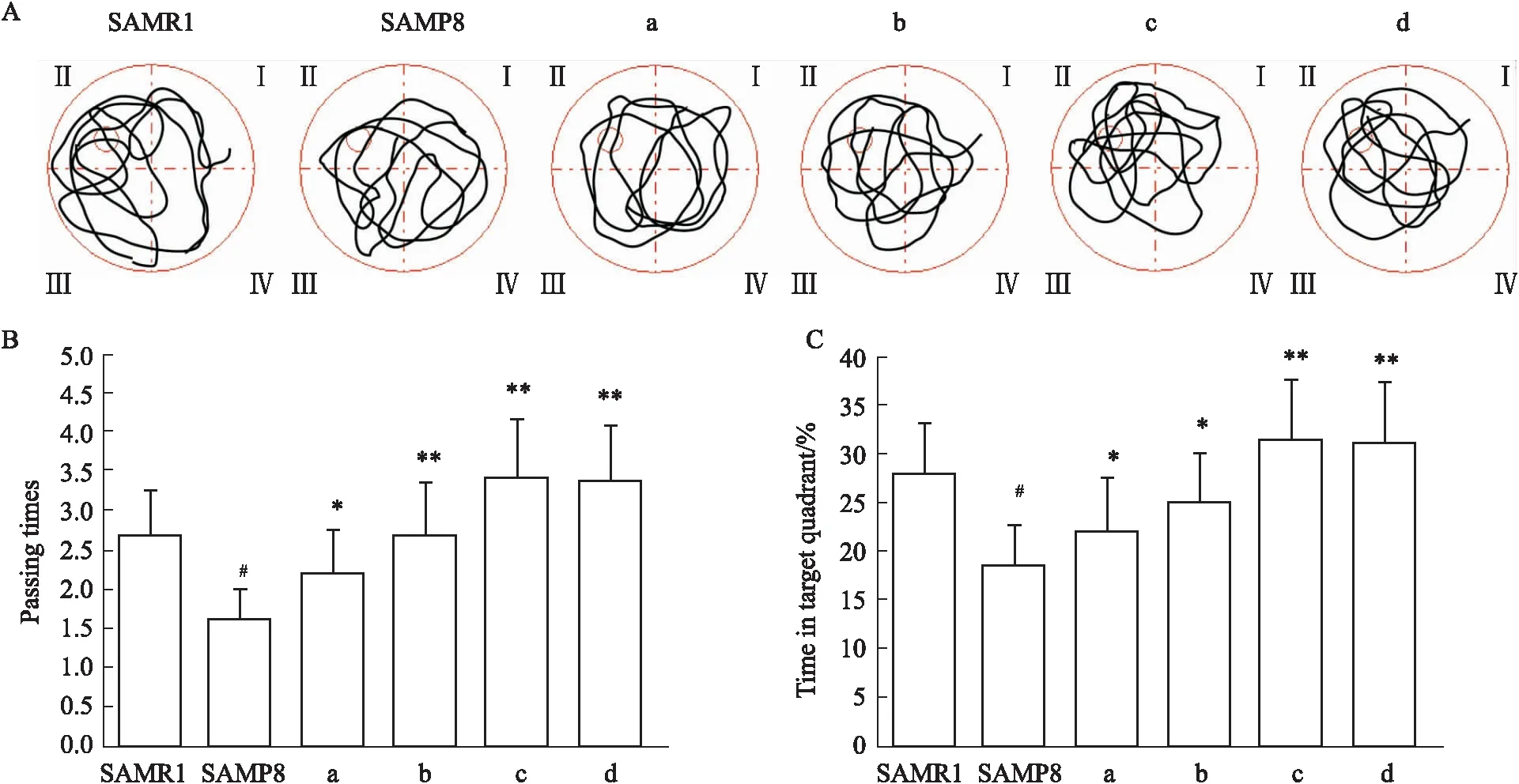
Fig 1 Spatial cognitive function of mice in each group detected by water maze test
2.2 HUK对SAMP8小鼠CA3区ChAT表达的影响免疫组化染色可见ChAT呈胞质胞膜染色,相较于SAMR1组,SAMP8组ChAT阳性细胞在CA3区表达明显减少;而HUK组ChAT阳性细胞在CA3区表达明显增多(Fig 2A)。随后,我们对各组ChAT的表达进行了rtPCR检测,SAMP8组表达明显低于SAMR1组(P<0.01),而给予HUK治疗后,ChAT表达明显高于SAMP8组(P<0.01),见Fig 2B。

Fig 2 Spatial cognition of each group detected by water maze test(× 400)
2.3 HUK对氧化应激水平的影响由Fig 3可见,与SAMR1组比较,SAMP8组小鼠CA3区中IL-1β、IL-18含量及MPO活性明显升高(P<0.01)。与SAMP8组比较,HUK明显降低小鼠CA3中IL-1β、IL-18含量及MPO活性(P<0.01)。以上结果提示HUK能降低SAMP8小鼠中氧化应激水平。

Fig 3 Effect of HUK on level of oxidative stress##P<0.01 vs SAMR1 group; **P<0.01 vs SAMP8 group.
2.4 HUK对SAMP8小鼠突触相关蛋白表达的影响为进一步验证HUK能否改善SAMP8小鼠的突触可塑性,我们采用Western blot法检测了CA3区突触相关蛋白PSD95和SYN。与SAMP8小鼠比,SAMR1小鼠PSD95和SYN蛋白表达量均低(P<0.01);而经HUK干预7 d后,PSD95和SYN蛋白的表达均增加(P<0.01),见Fig 4。本部分结果表明HUK改善了SAMP8小鼠海马突触可塑性。

Fig 4 Effect of HUK on expression of synapse-related proteins ##P<0.01 vs SAMR1 group; **P<0.01 vs SAMP8 group.
2.5 HUK对pCREB-BDNF信号通路的影响Western blot分析显示,与SAMR1组相比,SAMP8小鼠BNDF和pCREB水平明显降低(P<0.01)。而SAMP8小鼠经HUK处理后,BNDF和pCREB水平显着增加(P<0.01)。见Fig 5。以上结果表明HUK增强了SAMP8小鼠海马中的pCREB/BDNF信号传导,这可能是HUK保护突触可塑性的潜在机制。
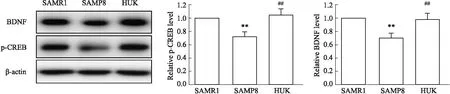
Fig 5 Effect of HUK on pCREB-BDNF signaling pathway##P<0.01 vs SAMR1 group; **P<0.01 vs SAMP8 group.
3 讨论
本研究表明,HUK能明显改善8月龄SAMP8小鼠的空间认知功能,HUK提高了SAMP8小鼠CA3区ChAT的表达,并且HUK能减轻小鼠脑组织中氧化应激水平,促进突触相关蛋白的表达。
近年来研究发现,HUK具有多方面的药理活性,能够减弱氧化应激和炎症反应,减少神经元凋亡,并保护濒临坏死的神经元[4-5]。临床研究证实,HUK能够减轻神经功能损伤,促进脑组织修复能力,促进神经营养因子的表达,具有潜在的改善认知功能作用[4]。但目前尚无关于HUK与SAD模型中神经元细胞ChAT、突触蛋白表达的研究报道。
AD的标志性病理学改变包括:由Aβ构成的淀粉样炎性斑块、Tau蛋白过度磷酸化构成的神经原纤维缠结。少部分AD患者与Aβ前体蛋白和早老素1、2基因突变有关,大多数AD患者呈散发性,与突变基因无关联。SAD的多种致病因素可能最终导致Aβ的瀑布效应,意味着在Aβ形成之前SAD患者存在其他形式病理改变[6]。乙酰胆碱是中枢神经系统最重要的神经递质之一,在学习记忆中起重要作用。胆碱乙酰转移酶(ChAT)为乙酰胆碱生成的限速酶。AD患者多处脑区ChAT活性降低,表达量减少,与患者痴呆程度、脑内Aβ含量呈负相关。
SAMP8小鼠模型是由AKR/J小鼠品系基于自然衰老筛选而来的快速老化模型,未经基因突变修饰,表现出记忆力和学习能力的迅速恶化[7-8]。SAMP8小鼠具有AD早期的多种特征,例如神经元和树突棘的丧失、胆碱能缺陷、氧化应激增加等[3,7],因此,SAMP8是进行SAD研究的良好动物模型。本研究中,SAMP8小鼠空间认知能力较SAMR1小鼠明显下降,并且出现ChAT减少、氧化应激水平增加,进一步证实了以上结论。AD发病机制中的一个关键环节是促炎细胞因子增加为特征的神经炎性相关氧化应激过程,IL-1β和IL-18介导谷氨酸兴奋毒性与氧化应激反应,导致神经细胞死亡。IL-1β和IL-18参与了神经损伤后炎性小体介导的氧化应激作用,去氧化状态后,IL-1β、IL-18、cleaved-caspase-1和MPO表达降低[9-10],而氧化应激还可进一步增加IL-1β和IL-18的产生[11]。Corbo等[12]最新研究表明,白细胞端粒长度和IL-1β和IL-18是认知障碍转变为AD的新型标志物,AD不仅是神经退行性疾病,而且是具有加速氧化应激水平的“全身”疾病。AD患者及动物模型中氧化应激水平增高,而神经元内氧化应激会导致突触减少及树突棘丢失[13]。在1-2月龄APP/PS1小鼠中Aβ斑周围存在较强氧化应激水平,而突触明显减少,在3xTg-AD小鼠的研究中发现海马区域明显氧化应激水平,海马神经元传递信号级联降低,并影响突触可塑性[14-15]。
胆碱能神经元主要分布于海马CA3区,而海马CA3区与学习及记忆密切相关,CA3区损伤会出现学习和记忆功能障碍。研究显示SAD患者CA3区出现明显增龄性退变[16]。海马区突触相关蛋白也参与了学习记忆过程,突触素(SYN)是突触前末端标记物,而突触后密度蛋白95(PSD95)则是突触后末端标记物。除了神经营养功能外,脑源性神经营养因子(BDNF)还可以有效调节突触可塑性、学习和记忆。突触蛋白的表达和活性通过BDNF来调节,海马中SYN和PSD95的表达会通过增强的BDNF信号传导而增加[17],而BDNF的神经保护作用则依赖于磷酸化转录因子环磷腺苷反应元件结合蛋(pCREB)介导的转录[18]。本研究中,SAMP8小鼠突触相关蛋白及BDNF、pCREB均减少,与上述研究结果相一致。
综上,本研究证明HUK具有提高SAD动物模型认知功能的作用,能改善CA3区神经元功能,其机制可能通过促进CA3区ChAT表达、减轻氧化应激水平及增加突触相关蛋白表达来实现。由此初步阐明HUK具有潜在的AD治疗效果。在以后的工作中,我们还将继续通过体外细胞实验,以进一步阐明HUK的分子作用机制。
猜你喜欢
作文周刊·小学二年级版(2022年20期)2022-05-05 01:33:06
世界科学技术-中医药现代化(2020年2期)2020-07-25 02:05:56
创新作文(小学版)(2019年10期)2019-09-25 08:12:28
中国组织化学与细胞化学杂志(2017年1期)2017-06-15 20:27:42
中成药(2017年6期)2017-06-13 07:30:35
小学生学习指导(低年级)(2017年5期)2017-05-04 04:14:38
西南军医(2016年6期)2016-01-23 02:21:19
吉林大学学报(医学版)(2015年4期)2015-12-17 07:48:13
作文与考试·小学高年级版(2015年17期)2015-05-30 10:48:04
癌变·畸变·突变(2015年3期)2015-02-27 06:15:12
