
高效液相色谱法测定土壤中16 种多环芳烃
2022-12-21 12:13高铮
环境保护与循环经济 2022年10期
高铮
(河北众淳环境检测技术有限公司,河北 石家庄 050000)
1 引言
多环芳烃(PAHs)作为一种分子中含有2 个或2个以上苯环的芳香族化合物,由煤炭、石油等有机物的不完全燃烧产生,分子结构中具有稳定的共轭体系[1],在一定条件下进入大气、水源、土壤等环境介质后很难降解,容易在生物体内积累,当其进入身体,将直接影响接触人的身体健康,甚至会导致癌症等恶性疾病的发生,因此研究多环芳烃的测定具有非常重要的意义[2]。
由于土壤成分的复杂性,多环芳烃不经前处理而直接定量测定会产生许多干扰,测定前有必要对多环芳烃进行前处理。本文采用索氏提取、固相萃取净化,再经高效液相色谱法测定,建立一种土壤样品中多环芳烃的定量分析方法。
2 实验
2.1 试剂与仪器
2.1.1 试剂与材料
乙腈,液相色谱级(Fisher Chemical);二氯甲烷、正己烷、丙酮,液相色谱级(科密欧);石英砂,AR(国药集团化学试剂);无水硫酸钠,GR(大茂化学试剂);乙腈中16 种多环芳烃标准溶液(坛墨质检标准物质中心);土壤样品采集自大名县隆丰牧业厂区内,取土深度为15 cm,经测定土壤样品干物质含量(ω)为92.1%;十氟联苯标准溶液。
2.1.2 仪器设备
氮气吹扫仪(杭州聚同电子);电热恒温水浴锅(天津市泰斯特仪器);索氏提取器;LC-16 型高效液相色谱仪(日本岛津);C18 色谱柱;Florisil 柱:1 g/6 mL(Agela Technologies)。
2.2 方法
2.2.1 索氏提取
称取10 g 土壤样品,与无水硫酸钠混合并研磨。参考HJ 748—2016《土壤和沉积物 多环芳烃的测定高效液相色谱法》规定的多环芳烃的提取条件,调节水浴锅温度75~85 ℃后,将制备好的样品放入滤纸筒中,加入二氯甲烷—丙酮混合提取剂,连续回流提取[3],18 h 后将提取完成后的提取液通过一薄层的无水硫酸钠过滤到氮吹管中,室温下氮吹浓缩转化溶剂为正己烷,定容至1.0 mL[4]。
2.2.2 固相萃取
将Florisil 柱固定于固相萃取仪上,参考HJ 748—2016 多环芳烃的固相萃取净化条件,经二氯甲烷预冲洗、正己烷平衡后,将提取液加到Florisil柱中,其间用正己烷冲洗浓缩所用氮吹管,洗涤液一并加到Florisil 柱中。用二氯甲烷与正己烷混合溶液洗脱,浸润5 min 后收集洗脱液于氮吹管中,室温下氮吹浓缩至1 mL,加入乙腈继续氮吹至溶剂完全转化为乙腈,最终定容至1.0 mL,密封保存留待液相色谱分析[5]。
3 结果与讨论
3.1 色谱条件选择
3.1.1 检测器及波长的选择
经实验,使用荧光检测器测定时,苊烯及替代物十氟联苯在荧光谱图中不出峰,故选择紫外检测器进行16 种多环芳烃的测定。根据液相色谱仪紫外检测器波长扫描,最大紫外吸收波长见表1[6]。

表1 多环芳烃各组分最大紫外吸收波长nm
根据16 种多环芳烃最大紫外吸收波长,选择220 nm 作为实验检测波长,目标组分的分离度最佳且峰面积的响应最适中,还能够降低杂峰的干扰。
3.1.2 流动相的选择
多环芳烃各组分具有相似的分子结构和性质,在色谱柱的保留时间也比较接近,需要使用梯度洗脱更有效地分离多环芳烃16 个组分。梯度洗脱程序见表2。

表2 梯度洗脱程序
3.1.3 流动相流速的确定
选择在4 种不同流速下测定多环芳烃标准物质,出峰情况见表3。

表3 不同流速下出峰情况 mL/min
根据测定结果,选择流动相流速1.20 mL/min 进行HPLC 分析,此时峰分离谱图效果最佳。
3.2 定性与定量分析
以多环芳烃标准物质的保留时间定性、峰面积定量,得到多环芳烃的标准色谱图,见图1。
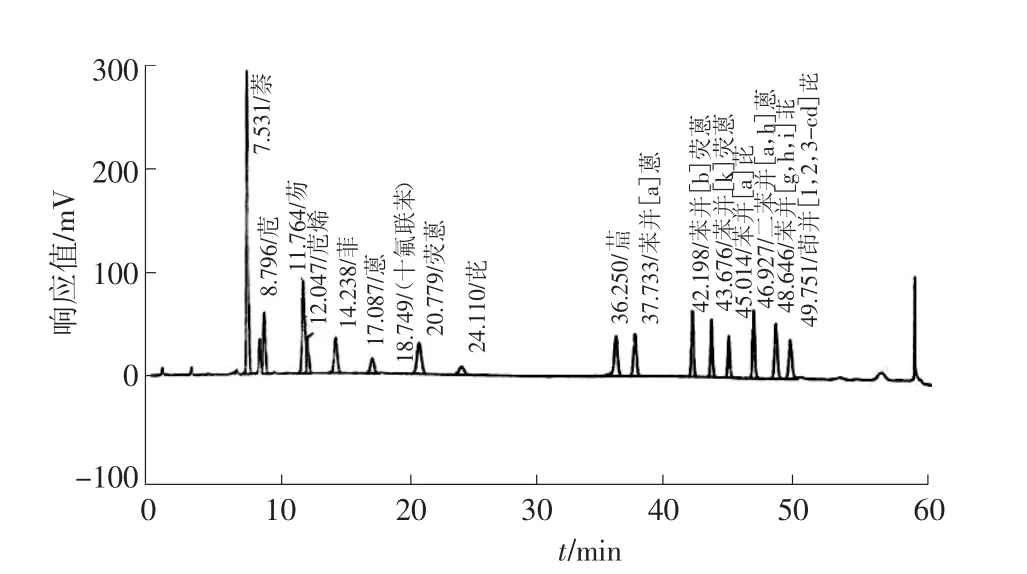
图1 多环芳烃标准色谱图
3.3 标准曲线绘制
稀释乙腈中16 种多环芳烃标液,制备标准浓度分别为0.04,0.10,0.50,1.00,2.00,5.00 μg/mL 的标准样品,将标准系列注入液相色谱仪,根据HPLC 分析条件分离检测,得到多环芳烃标准色谱图,绘制以浓度为横坐标、峰面积为纵坐标的多环芳烃标准曲线[7],见表4。

表4 回归方程及相关系数
3.4 质量控制
每个样品需在进行前处理之前加入一定含量的替代物十氟联苯,再按照前处理步骤依次进行,十氟联苯回收率需保证在60%~120%之间,超出十氟联苯回收率范围的样品视为无效。
3.5 检出限
根据HJ 168—2020《环境监测分析方法标准制订技术导则》附录A.1.1 中多组分目标化合物检出限的确定方法,分别取7 组10.0 g 石英砂作为空白试样,向其中加入20.0,10.0 μg/mL 的多环芳烃标准物质(加标量0.20 μg)。按照本方法经样品提取、浓缩、净化,最终将溶剂转化成乙腈,并准确定容至1.00 mL待测。检出限计算公式为[8]:

16 种多环芳烃检出限见表5。

表5 16 种多环芳烃检出限 μg/kg
3.6 精密度
向10.0 g 空白石英砂中分别加入含量为0.20,0.50,1.00 μg 的16 种多环芳烃标准物质,以代替实际样品做精密度测定。按照本方法经样品提取、浓缩、净化,最终将溶剂完全转化成乙腈,并准确定容至1.00 mL 待测。每个加标量按照相同步骤进行6次平行试验,结果见表6。

表6 16 种多环芳烃精密度
3.7 准确度
取10.0 g 厂区内土壤样品(取土深度为15 cm、干物质含量为92.1%),加入含量为1.00 μg 的多环芳烃标准物质做准确度测定,同时取10.0 g 样品不加标测定样品含量,按照本方法经样品前处理,溶剂转化成乙腈,并准确定容至1.00 mL 待测。按照相同步骤进行测定并计算加标回收率[5],结果见表7。

表7 16 种多环芳烃准确度
4 结论
通过索氏提取、固相萃取净化,使用高效液相色谱紫外检测器定量分析,建立一种检测土壤中16 种多环芳烃的方法,通过实验测定土壤样品,加标回收率为67.0%~79.4%。由于土壤样品基体的复杂性,实验测定存在较多干扰,加标回收率并不是很高,未来应加强实验探究,通过改进前处理提取浓缩净化方式,减少实验过程中多环芳烃的损失,提高加标回收率。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
波谱学杂志(2022年2期)2022-06-14
石油炼制与化工(2022年2期)2022-02-15
科学家(2021年24期)2021-04-25
化工管理(2020年26期)2020-10-09
山东化工(2019年2期)2019-02-21
电子技术与软件工程(2016年24期)2017-02-23
橡胶工业(2015年9期)2015-08-29
橡胶工业(2015年6期)2015-07-29
橡胶工业(2015年4期)2015-07-29
