
一例家族性醛固酮增多症II型合并WFS1基因突变的诊疗和基因检测分析
2022-11-29 09:20孙致连何俊莹程筱玲谭晓霞吴伟华
遗传 2022年11期
孙致连,何俊莹,程筱玲,谭晓霞,吴伟华
遗传资源
一例家族性醛固酮增多症II型合并基因突变的诊疗和基因检测分析
孙致连,何俊莹,程筱玲,谭晓霞,吴伟华
深圳市罗湖区人民医院内分泌科,深圳 518001
原发性醛固酮增多症(primary aldosteronism, PA)又简称原醛症,是指肾上腺皮质分泌过量醛固酮(aldosterone,ALD),导致水钠潴留及肾素–血管紧张素系统受抑制,从而引起高血压、低血钾的一类疾病。家族性醛固酮增多症II型(familial hyperaldosteronism type II, FH-II)是PA的少见病因,为常染色体显性遗传病。本文收集1例自2014年起病以来反复高血压、低血钾的病例,该患者诊断结果一直未明确。2021年通过基因检测证实该患者存在及基因突变,其母亲为杂合携带,父亲为野生型。结合内分泌功能试验结果及影像学资料,考虑患者为FH-II,同时合并基因突变。通过总结分析该病例特点及基因检测结果,对于临床难以明确病因的PA建议可行基因测序。该病例也为后续遗传学研究提供临床资料。
家族性醛固酮增多症II型;;
原发性醛固酮增多症(primary aldosteronism, PA),简称原醛症,是继发性高血压的常见原因,以高血压、低血钾为主要特点,发病年龄高峰30~50岁,女性多见。PA患者心脏、肾脏等高血压靶器官受损严重程度较原发性高血压患者更为严重,早期诊疗尤为重要。家族性醛固酮增多症(familial hyperaldosteronis,FH)是PA的少见病因之一,根据目前已知的致病基因,分为I型(FH-I)、II型(FH-II)、III型(FH-III)、IV型(FH-IV),均为单基因常染色体显性遗传病。FH-II存在突变,此外有研究发现FH-II还与染色体7p22区域相关,但具体的发病机制目前尚不明确[1,2]。本文主要报道了1例发病7年后通过内分泌功能试验、基因检测确诊的FH-II合并基因变异病例。对该例病例特点进行总结分析,旨在为后续该疾病的遗传学研究提供临床资料,同时为临床医师明确这类疾病的诊断提供思路。
1 对象与方法
1.1 对象及临床资料收集
患者37岁女性,因血压控制不佳、低血钾于2021年就诊于我院,收集患者的病史、体格检查、实验室检查、影像学检查,以及患者及其父母基因检测等临床资料。本研究获得深圳市罗湖区人民医院伦理委员会审核批准。患者及其父母均签署知情同意书。
1.2 全外显子测序
利用序列捕获技术将全基因组区域DNA捕捉并富集,然后进行高通量,基因测序由金域检验中心完成。
1.3 序列分析验证与解读
应用了xGen全外显子组panel进行DNA建库,进行分析时,以KMTD/KMED、HGMD、ClinVar、ESP6500、G1000、dbSNP为主要对照的突变/变异数据库。应用Illumina二代测序仪进行测序检测。并对其父母进行了针对所检出变异位点的亲属一代测序验证。根据美国医学遗传学和基因组学学会(American College of Medical Genetics and Genomics, ACMG)遗传变异分类标准将检测到的变异分类为“致病”、“可能致病”和“意义不明确”。
2 结果与分析
2.1 患者病史
问诊患者既往病史,患者2014年体检发现血压升高,约210/110 mmHg,无头晕、呕吐,无肢体乏力,无头痛、心悸、出汗,无满月脸、水牛背。外院完善尿蛋白、肾功能、甲功、醛固酮/肾素活性比值(aldosterone-to-renin activity ratio, ARR)、肾动脉、肾上腺CT未见异常,外院查血钾3.02mmol/L,诊断“高血压、低血钾查因”,予“硝苯地平控释片30 mg bid、螺内酯片 80mg qd”治疗,用药后血压降至150/80 mmHg左右,血钾恢复正常。后患者自行停用螺内酯,减少硝苯地平控释片用量。2015年因头晕再次外院就诊,测血压163/108 mmHg,随机血糖约12 mmol/L,血钾浓度3.18mmol/L,同步尿钾浓度 36.6 mmol/24 h (当血钾浓度<3 mmol/L时,尿钾浓度>20 mmol/24 h为排出增多;当血钾浓度<3.5 mmol/L时,尿钾浓度>25 mmol/24 h为排出增多),ARR、儿茶酚胺、肾上腺CT未见异常,诊断“特发性醛固酮增多症?和2型糖尿病”,出院后长期予“硝苯地平控释片30mg qd、螺内酯80 mg qd、二甲双胍0.5 g tid”治疗,平素血压140/80 mmHg左右,未定期监测血钾,血糖控制情况不详。因拟行高血压病因筛查,患者停用螺内酯4周后,于2021年12月来我科住院。既往有高脂血症及高尿酸血症病史。已婚未育。父母非近亲结婚。母亲及外祖父有高血压病病史,母亲约40岁发现血压升高,外祖父发病年龄不详,血压均不易控制。否认外祖母、祖父母、父亲、妹妹高血压。入院查体:血压166/114mmHg,身高162cm,体重80kg,体重指数30.48 kg/m2。体型肥胖,无多毛、紫纹、黑棘皮,无痤疮、满月脸、水牛背,听力正常,甲状腺无肿大,无骨骼增大增粗、唇舌增厚,肺心腹查体未见异常,双下肢无浮肿。
2.2 患者临床检查结果
患者自2021年住院于我科后,对其进行了较为全面的生理生化和影像学检查。肾功能、尿蛋白、ANCA、ANA、甲状腺功能、性激素、人绒毛膜促性腺激素、人体生长激素及IGF-1均在正常范围。糖尿病抗体阴性。骨代谢指标降低。泌尿系彩超、心脏彩超、肾动脉及腹主动脉彩超均未见异常。血气分析显示酸碱度7.46,钾离子2.8mmol/L。连续3次留取患者24 h尿电解质,以及同步血电解质提示在低血钾状态下尿钾排出增多(表1)。口服葡萄糖耐量试验(oral glucose tolerance test, OGTT)及胰岛素/C肽释放试验,结果提示患者存在高胰岛素血症(表2)。对其进行PA初筛试验,卧位高血压5项显示肾素活性(plasma renin activity,PRA)1.90 μg/L/h (正常参考值:卧位0.15~2.33 μg/L/h,立位1.31~ 3.95 μg/L/h)、血管紧张素II 1.0 μg/L(正常参考值:卧位25~129 μg/L,立位49~252 μg/L)、醛固酮(aldosterone,ALD)156.58 ng/L(正常参考值:卧位10~160 ng/L,立位40~310 ng/L)、ARR 8.24(正常参考值:0~30),提示患者ALD水平正常值高限,PRA正常,ARR未达切点值。随后进行PA确诊试验:(1)卡托普利试验,在坐位1 h后口服50 mg卡托普利,服药前及服药后1 h、2 h测定血浆PRA及血ALD水平,结果提示ALD不受抑制(表3); (2)生理盐水试验,在卧床休息1 h后,4 h静滴2 L 0.9%氯化钠溶液,在输注前及输注后分别采血测PRA、ALD水平,结果提示ALD不受抑制(表4)。经临床试验初步确诊PA后,进一步完善肾素–血管紧张素–醛固酮系统(renin-angiotensin-aldosterone system, RAAS)体位试验,体位试验结果提示患者在立位时ALD水平升高(表5)。此外,还对患者的肾上腺其他激素进行了评估,结果显示:甲氧基肾上腺素类、儿茶酚胺、硫酸脱氢表雄酮、雄烯二酮、17-羟孕酮含量均在正常范围内;皮质醇/促肾上腺皮质激素(adrenocorticotrophic hormone, ACTH)昼夜节律正常,1 mg地塞米松抑制试验(1 mg dexamethasone suppression test,1mgDST)可使皮质醇被抑制(表6)。对患者进行肾上腺CT平扫,结果提示:左侧肾上腺外侧肢似见结节影,与肾上腺密度相仿,右侧肾上腺形态位置大致如常(图1)。

表1 连续3次24 h血尿同步电解质

表2 OGTT及胰岛素/C肽释放试验

表3 卡托普利试验
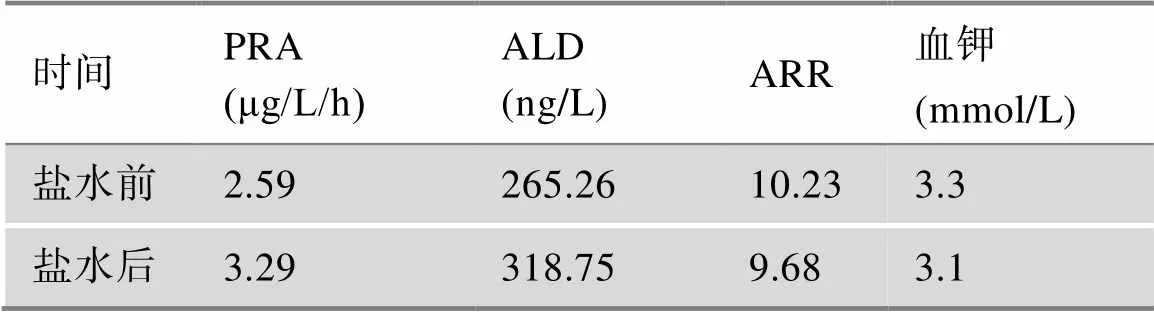
表4 生理盐水试验

表5 肾素–血管紧张素–醛固酮系统体位试验评估
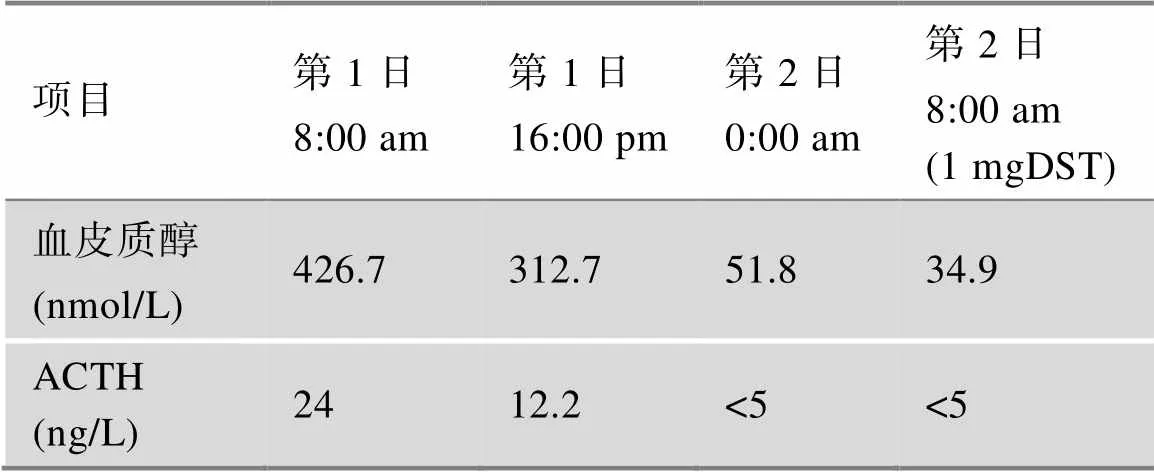
表6 皮质醇/ACTH昼夜节律+午夜1 mgDST
2.3 基因检测结果分析
对患者行遗传病全外显子组测序,对其父母进行针对患者所检出变异位点的测序,发现患者及其母亲存在基因(NM_004366)杂合变异:c.1327-17C>A(本变异位于内含子区域,虽越来越多的研究证明内含子区的变异可能通过影响mRNA的剪切、修饰、翻译效率等参与疾病的发生发展,但直接对蛋白质翻译的影响不明确),位于该基因12号内含子区(图2,A~C)。另外还检测到先证者及其母亲存在基因(NM_006005.3)杂合变异:c.2144G>T (p.Ser715Ile),位于8号外显子区(图2, D~F)。其父亲未检测到基因变异。
3 讨论
PA是继发性高血压的常见原因,因知晓率低,仍有很多患者未被及时、准确的诊断。2010年由中华医学会内分泌学分会牵头,对全国11个省19个中心的1656例难治性高血压患者进行了流行病学调查,其中PA的患病率为7.1%[1]。由此可见,PA是一种被忽视的常见病。与原发性高血压患者相比,PA患者的高血压相关靶器官损害更为严重,更易发生心血管事件,因此早期诊断及治疗尤其重要。PA典型的临床表现为高血压和低血钾,高血压为最常见的表现,部分患者血钾可正常,临床上亦有单纯以低血钾为表现的案例报道。PA根据病因不同可分为6型,FH是其中之一,目前FH又分为I~IV型,FH-II占PA比例<6%[1,2]。
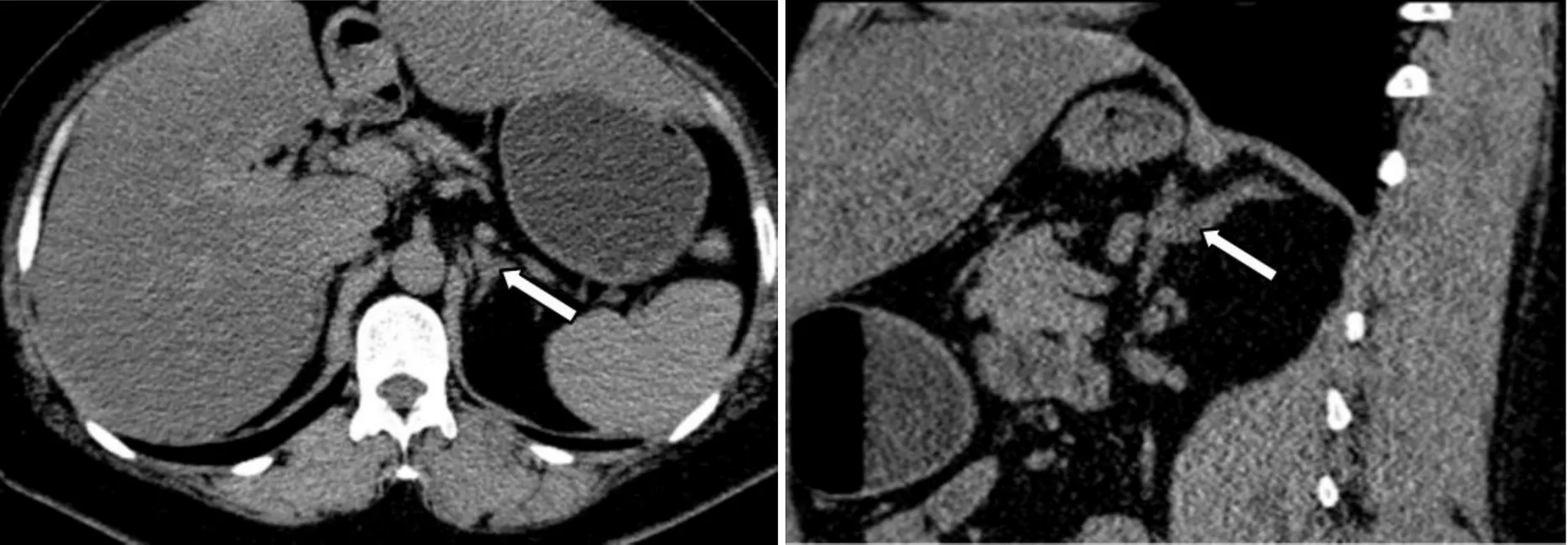
图1 患者的肾上腺平扫CT影像
左图为横断面,右图为矢状面,箭头指向左侧肾上腺外侧肢结节影。
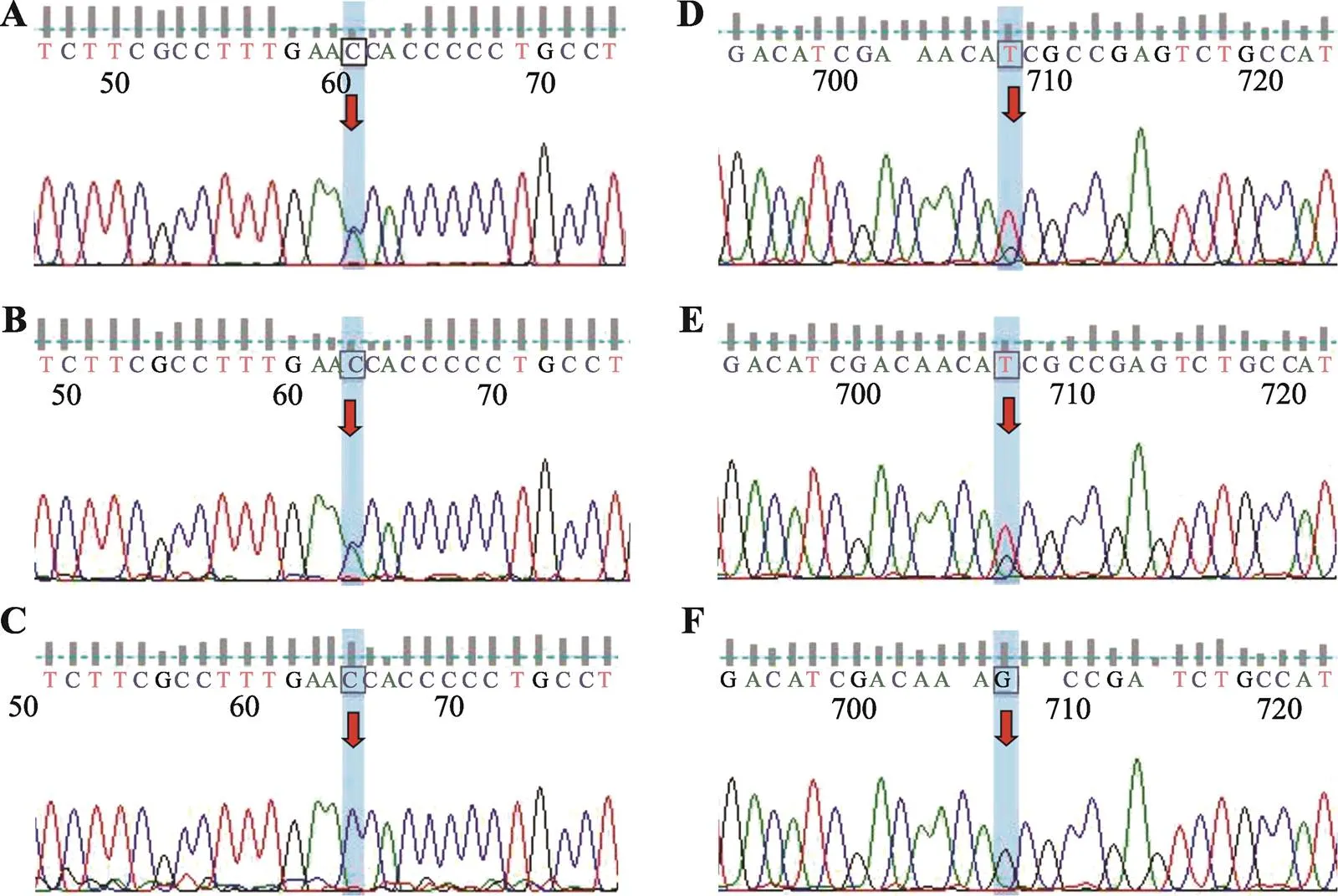
图2 患者及其父母的基因检测结果分析
A~C为基因位点,A和B变异位点:NM_004366:c.1327-17C>A(p.?),C为无变异野生型(阴性对照)。D~F为基因位点,D和E变异位点:NM_006005.3:c.2144G > T(p.Ser715Ile),F为无变异野生型(阴性对照)。A、D为先证者,B、E为其母亲,C、F为其父亲。
本例患者30岁左右出现血压明显升高,合并有无症状性自发低血钾、肾性失钾,临床应高度怀疑PA。行PA初筛试验,PRA正常范围,ALD正常值高限,ARR正常,初筛阴性。但ARR受许多因素的影响,如年龄、性别、饮食、血钾、药物、采血方法等,本例患者有低钾血症,存在致ARR假阴性因素存在,且临床高度怀疑PA,进一步行确诊试验。卡托普利试验和生理盐水试验,均提示ALD不能被抑制,支持PA诊断。再进行分型诊断,患者2014年肾上腺CT未见异常,2021年复查肾上腺CT提示左侧肾上腺结节样增生可能,基因检测提示存在基因变异,染色体位置:Chr.3: 184072778,变异位点位于该基因12号内含子区。先证者父母该变异位点基因检测提示母亲为杂合携带,父亲未检测到变异基因。该基因dbSNP147数据库有收录(rs752807616),根据ACMG指南,变异评级为意义不明变异(PP2+PP4),但结合患者临床表现、辅助检查结果,本团队仍然考虑基因变异具有致病性。越来越多的研究表明内含子是真核生物基因组的重要组成部分。内含子可以通过影响转录速率、核输出和转录稳定性来提高转录水平,此外,内含子还可以提高mRNA翻译的效率[3]。该患者基因内含子区域发生变异,可能通过影响基因转录及蛋白翻译等,参与FH-II的发生发展,但仍需后续发现更多家谱系证明其临床意义。综上,患者高血压、低血钾、高醛固酮、左侧肾上腺结节样增生、存在致病基因,已排除库欣氏综合征和嗜铬细胞瘤,结合患者早发高血压家族史、母亲存在致病基因,故考虑FH-II诊断。因FH-II目前尚缺乏确切的治疗经验,在手术及药物治疗方式选择上需权衡利弊。本例患者经过醛固酮拮抗剂螺内酯20 mg qd,以及硝苯地平控释片30 mg qd降压、氯化钾缓释片1 g tid补钾治疗后血压、血钾可维持在正常范围,后续随访患者未再发生严重低血钾、高血压以及血压大波动。
在大多数家系中,FH-II垂直传播,常提示为常染色体显性遗传疾病,具体机制尚未明确。2018年的两项研究均证实基因变异与FH-II相关[4,5]。编码电压门控氯离子通道,该通道位于肾上腺皮质球状带细胞膜,基因变异可使该通道开放频率增加,促使醛固酮合成限速酶表达增加,使醛固酮产生增加[4]。一项类固醇代谢组学研究[6]发现糖皮质激素分泌增多常见于PA,并导致相关的代谢风险增高,与对照组和亚临床库欣综合征患者相比,PA患者的糖皮质激素代谢物排泄量显著增加,其代谢风险与糖皮质激素正相关。该患者代谢综合征、骨吸收指标降低符合此特点。
一项涉及31个回顾性和前瞻性研究的荟萃分析结果显示:PA患者的糖尿病患病风险较原发性高血压患者增加,23.0%~35.3%的PA患者会伴发糖尿病或糖耐量异常,其机制可能为高醛固酮水平影响胰岛β细胞功能和靶组织对胰岛素的敏感性[7]。PA患者以餐后血糖升高为主,空腹血糖升高不明显[8]。该患者血糖升高特点与此相符。另外在本次基因检测中还发现先证者及其母亲存在基因突变。基因如发生致病性变异最常引起Wolfram综合征,典型特征是糖尿病、视神经萎缩、尿崩症和耳聋,其他临床特征可能包括肾脏异常、共济失调、痴呆或智力低下,以及各种精神疾病[9]。此外,基因变异还可引起非Wolfram综合征性糖尿病。有研究表明中国1型糖尿病患者中基因变异同基因变异一样常见,估计自身抗体阴性的1型糖尿病患者中有5%因基因变异而患有非Wolfram综合征性糖尿病,且隐性基因变异并不少见[10]。目前的研究中,基因变异还参与2型糖尿病的发病过程。至今至少有70个基因被认为与2型糖尿病的发生发展有关。其中,、、和基因变异是该病发病机制中最常报道的[11]。而且,不同易感基因在人群的发病率有所差异[12]。基因编码多种蛋白,如wolframin蛋白,该蛋白有助于胰岛素的生物合成和血糖的调节,还可调节胰高血糖素样肽-1 (GLP-1)表达[13]。这些基因变异在环境因素的作用下,如肥胖和超重,最终导致2型糖尿病。该患者存在基因变异,染色体位置:Chr.4:6303666,位于该基因8号外显子,dbSNP147数据库有收录(rs772022154),ACMG指南变异评级为可能致病的(PM2+PP1+PP2+PP3+PP5)。考虑基因可能参与糖尿病的发生发展,但本例患者病情复杂,糖尿病是由PA并发还是由基因变异致病,亦或两者共同参与目前尚不清楚。
本例患者长达7年之久才被确诊FH-II,提示对于临床高度可疑PA的患者,即使ARR阴性也建议进行确诊试验,降低漏诊率。FH-II从临床特征和生化检查方面很难与散发的醛固酮增多症区别开来,故需询问是否有肾上腺腺瘤或增生所致的PA家族史,还应做基因检测以确诊。本例先证者及其母亲基因检测中均发现同时存在基因与基因变异,这是以往未曾报道的。本例患者早发高血压、糖尿病,考虑基因与基因变异可能分别参与PA及糖尿病的发生发展。目前先证者无听力、尿崩及神经精神疾患,后期需继续随访,对其后代基因情况以及临床表现也需随访。本例中先证者早发高血压、糖尿病,基因与基因的变异对疾病进程、靶器官损害是否有互相促进作用,有待后续更多该基因变异位点的发现、统计及分析。
[1] Chinese Society of Endocrinology. Expert consensus on the diagnosis and treatment of primary aldosteronism (2020)., 2020, 36(9): 727–736.
中华医学会内分泌学分会. 原发性醛固酮增多症诊断治疗的专家共识(2020版). 中华内分泌代谢杂志, 2020, 36(9): 727–736.
[2] Wang HP, Tong AL. Research progress on gene mutation related primary aldosteronism., 2021, 41(2): 87–90.
王慧萍, 童安莉. 原发性醛固酮增多症相关的基因突变研究进展. 国际内分泌代谢杂志, 2021, 41(2): 87–90.
[3] Shaul O. How introns enhance gene expression., 2017, 91(Pt B): 145–155.
[4] Scholl UI, Stölting G, Schewe J, Thiel A, Tan H, Nelson- Williams C, Vichot AA, Jin SC, Loring E, Untiet V, Yoo T, Choi J, Xu SX, Wu AH, Kirchner M, Mertins P, Rump LC, Onder AM, Gamble C, Mckenney D, Lash RW, Jones DP, Chune G, Gagliardi P, Choi M, Gordon R, Stowasser M, Fahlke C, Lifton RP. CLCN2 chloride channel mutations in familial hyperaldosteronism type II., 2018, 50(3): 349–354.
[5] Fernandes-Rosa FL, Daniil G, Orozco IJ, Göppner C, El ZR, Jain V, Boulkroun S, Jeunemaitre X, Amar L, Lefebvre H, Schwarzmayr T, Strom TM, Jentsch TJ, Zennaro MC. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism., 2018, 50(3): 355–361.
[6] Arlt W, Lang K, Sitch AJ, Dietz AS, Rhayem Y, Bancos I, Feuchtinger A, Chortis V, Gilligan LC, Ludwig P, Riester A, Asbach E, Hughes BA, O'Neil DM, Bidlingmaier M, Tomlinson JW, Hassan-Smith ZK, Rees DA, Adolf C, Hahner S, Quinkler M, Dekkers T, Deinum J, Biehl M, Keevil BG, Shackleton CH, Deeks JJ, Walch AK, Beuschlein F, Reincke M. Steroid metabolome analysis reveals prevalent glucocorticoid excess in primary aldosteronism., 2017, 2(8).
[7] Wu VC, Chueh SJ, Chen L, Chang CH, Hu YH, Lin YH, Wu KD, Yang WS. Risk of new-onset diabetes mellitus in primary aldosteronism: a population study over 5 years., 2017, 35(8): 1698–1708.
[8] Hanslik G, Wallaschofski H, Dietz A, Riester A, Reincke M, Allolio B, Lang K, Quack I, Rump LC, Willenberg HS, Beuschlein F, Quinkler M, Hannemann A. Increased prevalence of diabetes mellitus and the metabolic syndrome in patients with primary aldosteronism of the GermanConn's Registry., 2015, 173(5): 665–675.
[9] Panfili E, Mondanelli G, Orabona C, Belladonna ML, Gargaro M, Fallarino F, Orecchini E, Prontera P, Proietti E, Frontino G, Tirelli E, Iacono A, Vacca C, Puccetti P, Grohmann U, Esposito S, Pallotta MT. Novel mutations in the WFS1 gene are associated with Wolfram syndrome and systemic inflammation., 2021, 30(3-4): 265–276.
[10] Li MH, Wang SH, Xu KF, Chen Y, Fu Q, Gu Y, Shi Y, Zhang M, Sun M, Chen H, Han XQ, Li YX, Tang ZK, Cai LJ, Li ZQ, Shi YY, Yang T, Polychronakos C. High prevalence of a monogenic cause in Han Chinese diagnosed with type 1 diabetes, partly driven by nonsyndromic recessive WFS1 mutations., 2020, 69(1): 121– 126.
[11] Yahaya TO, Salisu TF. A review of type 2 diabetes mellitus predisposing genes., 2019, 16(1): 52–61.
[12] Gong X, Zhang C, Yiliyasi A, Shi Y, Yang XW, Nuersimanguli A, Guan YQ, Xu SH. A comparative analysis of genetic diversity of candidate genes associated with type 2 diabetes in worldwide populations., 2016, 38(6): 543–559.
弓弦, 张超, 伊利亚斯·艾萨, 时瑛, 杨雪唯, 努尔斯曼古丽奥斯曼, 关亚群, 徐书华. 2型糖尿病易感候选基因在世界不同人群中的多样性比较分析. 遗传, 2016, 38(6): 543–559.
[13] Schäfer SA, Müssig K, Staiger H, Machicao F, Stefan N, Gallwitz B, Häring HU, Fritsche A. A common genetic variant in WFS1 determines impaired glucagon-like peptide- 1-induced insulin secretion., 2009, 52(6): 1075–1082.
Diagnosis, treatment and genetic analysis of a case of familial aldosteronism type II withgene mutation
Zhilian Sun, Junying He, Xiaoling Cheng, Xiaoxia Tan, WeihuaWu
Primary aldosteronism (PA) is a disease characterized by hypertension and hypokalemia due to the excessive aldosterone secretion from the adrenal cortex, which leads to the retention of both water and sodium, and the inhibition of the renin-angiotensin system as well. Familial hyperaldosteronism type II (FH-II) is known as an autosomal dominant hereditary disease, which is a scarce cause of PA. In this report, we cllected the clinical data of a patient with repeated hypertension and hypokalemia of uncertain diagnosis since 2014. Nevertheless, we discovered by genetic sequencing in 2021 that theandgene mutation of the patient, whose mother belongs to heterozygote genotype and father belongs to wild-type genotype. Combined with a series of endocrine function tests and imaging studies, the patient was finally certified her suffering from FH-II andgene mutation. By summarizing and analyzing the characteristics and genetic test results of this case, we recommended gene sequencing for patients with PA whose etiology is difficult to be determined clinically. This case also provides new clinical data for subsequent genetic studies of the disease.
familial hyperaldosteronism type II;;
2022-08-09;
2022-09-12;
2022-09-22
孙致连,硕士,副主任医师,研究方向:内分泌与代谢病方向。E-mail: sunzhilian@163.com
何俊莹,硕士,主治医师,研究方向:内分泌与代谢病方向。E-mail: hejunying1210@163.com
孙致连和何俊莹并列第一作者。
吴伟华,博士,主任医师,研究方向:糖尿病心肌病,血糖波动。E-mail: hui-90@163.com
10.16288/j.yczz.22-197
(责任编委: 周红文)
猜你喜欢
浙江医学(2022年14期)2022-11-28
中国人兽共患病学报(2022年9期)2022-10-19
科学导报(2021年29期)2021-06-03
中国生殖健康(2020年4期)2021-01-18
临床肝胆病杂志(2020年8期)2020-12-14
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
科海故事博览·下旬刊(2019年6期)2019-04-16
中国实用医药(2017年10期)2017-05-15
百科知识(2015年18期)2015-09-10
