
南-北方汉族人、韩国人和日本人遗传划分机器学习模型优化方案
2022-11-29 09:20孔永强刘金凯顾佳琪徐景怡郑雨诺魏以梁伍少远
遗传 2022年11期
孔永强,刘金凯,顾佳琪,徐景怡,郑雨诺,魏以梁,伍少远,
研究报告
南-北方汉族人、韩国人和日本人遗传划分机器学习模型优化方案
孔永强1,刘金凯1,顾佳琪2,徐景怡1,郑雨诺2,魏以梁2,伍少远1,2
1. 天津医科大学基础医学院生物化学与分子生物学系,天津市表观遗传学重点实验室,天津 300070 2. 江苏师范大学,江苏省系统发育与比较基因组学重点实验室,徐州 221116
中国汉族人、韩国人和日本人作为东亚主体人群,其中中国汉族人呈现由北向南的梯度混合,在遗传结构上存在不同程度的差异。为实现对中国南-北方汉族人、韩国人和日本人的高分辨率遗传划分,本研究收集和分析了文献报道和实验室前期数据筛选出的1185个东亚人群祖先信息性SNPs (ancestry informative SNPs, AISNPs),应用softmax与随机森林两种机器学习算法构建族群遗传划分模型,然后利用系统发育树、STRUCTURE和主成分分析方法进一步评估不同模型AISNPs位点组合的族群分类效果,最终筛选出234-AISNP的最优组合,softmax模型准确率为92%,实现了南方汉族人、北方汉族人、韩国人和日本人的高精度区分。本研究测试的两种机器学习算法模型为近距离人群的高分辨率划分提供了重要参考,可作为法医DNA族群推断体系位点开发的重要工具。
法医遗传学;祖先信息位点;机器学习;东亚人群;南北方汉族
在法医学案件侦破过程中,利用不同人群之间等位基因频率分布差异较大的遗传标记,即祖先信息标记(ancestry informative markers, AIMs),进行种族地域分析,可以缩小嫌疑人的侦查范围[1]。由于单核苷酸多态性(single nucleotide polymorphism, SNP)在人类基因组中含量丰富,常用于计算和筛选AIMs,被称为祖先信息性SNPs (ancestry informative SNPs, AISNPs)[2]。使用AISNP进行的群体遗传结构差异分析在医学全基因组关联研究和法医生物地理推断中发挥着重要作用[3]。在过去10年中,国内外已建立了多套常染色体AISNP检测分析系统,用于区分洲际或洲内的族群差异[4~7]。
目前基于DNA的种族地域推断研究已经从非、欧、东亚等洲际大人群的划分发展到对各自局部地区亚人群的精细划分[8,9]。中国汉族人、韩国人和日本人作为东亚主体人群,外形特征和语言文化极为相似,基因组研究表明这3个群体间的遗传成分存在细微差异[9,10]。中国汉族作为世界上最大的族群,其早期人类社会活动受到长江地理分隔作用的影响,人群遗传结构呈现由北向南的梯度混合模式[11]。本研究基于国内外对中国汉族、韩国和日本人群遗传结构研究的相关成果,集合了1185个AISNP[8~10,12~20],采用机器学习算法筛选SNP组合和建立高精度人群划分模型,以区分南-北方汉族人、韩国人和日本人。
1 材料与方法
1.1 样本收集
参考及测试样本数据为全基因组和芯片检测数据。参考数据集403份样本包括:来自千人基因组计划[21]的105份中国南方汉族样本、103份中国北京汉族样本、104份日本样本和来自韩国个人基因组计划[22]的91份韩国人样本(表1)。测试数据集199份样本包括:来自千人基因组新增[23]的58份中国南方汉族样本,来自人类基因组多样性计划[24]的10份中国北方汉族和27份日本样本,来自西蒙斯基因组多样性计划[25]的2份日本样本和2份韩国样本,以及来自亚洲多样性计划[26](分型基于Affymetrix genome-wide human SNP array 6.0芯片)的100份韩国样本(表2)。
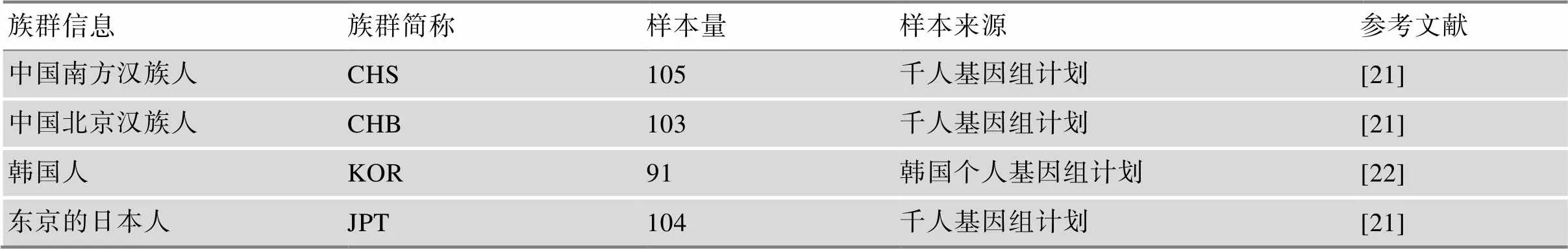
表1 参考集样本信息
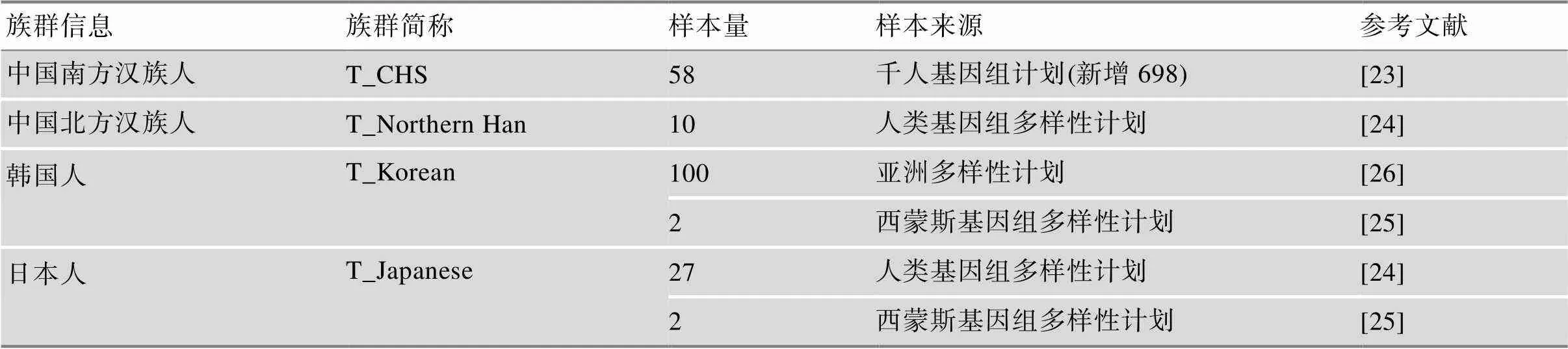
表2 测试集样本信息
1.2 SNP来源
前期研究和文献调研,从12篇文献[8~10,12~20]及实验室前期筛选数据[27,28]中收集到1185个AISNP (附表1)。文献研究的人群、数目及来源见表3。
1.3 质量控制
1.3.1 数据类型与处理
通过IBM Aspera v3.0.0[29]从IGSR数据库[30](https://www.internationalgenome.org/data-portal/sample)和韩国个人基因组计划[22](ftp://biodisk.org/Release/ KPGP/)下载了502份样本的全基因组数据,并且从中国科学院上海生命科学研究院徐书华课题组[26]获得了100份样本的芯片位点数据。所有样本的数据类型可分为fastq、cram与vcf格式。其中,参考和测试集中vcf格式数据是研究者分别通过GATK流程[22,30,31]和Birdsuite1.5.3[32]分析获得的。而测试集样本的fastq和cram格式数据是本课题组基于GATK4.1.9.0[33]流程进行质控、预处理和变异检测,从而获得不同数据库不同人群的vcf格式数据。
1.3.2 数据的质量控制
分别从参考及测试集样本的若干个vcf中提取1185个AISNP的基因分型数据,先去除样本基因分型缺失率大于10%的SNP,再去除SNP基因分型缺失率大于10%的样本,最终参考集、测试集数据含1128个AISNP,602份样本。
1.3.3 众数填充
测序过程的失误或其他原因会造成某些样本的某些SNP的基因分型是缺失即NN的状态,在共线性诊断及平均降准(mean decrease accuracy, MDA)交叉验证之前需对这些缺失的基因分型用R v4.0.2的imputeMissings v0.03包[34]进行众数填充,即用单人群中该SNP出现频率最多的基因分型填补。
1.4 基因分型及人群编码
由于SPSS共线性诊断及R v4.0.2中的softmaxreg v1.2[35]和randomForest v4.6-14[36]两个包要求变量为数值型形式,在众数填充后需要对基因分型的纯合和杂合按照0、1、2进行编码,人群则按照1-南方汉族、2-北方汉族、3-日本、4-韩国进行编码。

表3 AISNP收集与来源
1.5 系统发育树构建
利用R4.0.2中ggtree2.4.2包[37]以参考集、测试集样本基因分型数据构建系统发育树。
1.6 Hardy-Weinberg平衡及连锁不平衡分析
在进行SPSS共线性诊断及MDA交叉验证挑选SNP和模型评估之前,先进行Hardy-Weinberg平衡检验(Hardy-Weinberg equilibrium, HWE)[38]和连锁不平衡分析(linkage disequilibrium, LD)[39],通过设置不同的HWE和LDr阈值筛选AISNP组合。借助haploview v4.2[40]进行HWE和LD分析,其中HWE按照值(0~1)进行排序挑点,分别以0.05、0.01和这两个值的Bonferroni[41]校正值(0.05/1185、0.01/ 1185)为阈值;而LD按照r(0~1),r为1说明完全连锁,分别以0.8、0.5、0.2、0.1为阈值。
1.7 AISNP筛选
不同人群之间产生的遗传差异在很大程度上受到遗传漂变的影响,这就使得群体间差异位点即AISNP的选择显得尤为重要。通常研究者基于频率差异分析技术进行AISNP的筛选,如Wright’sF值[42]和I值[43](informativeness for assignment, Rosenberg’sIdivergence)。Wright’sF值(0~1)和I值(0~1)都是群体遗传学中衡量群体间分化程度的一个重要指标,其大小反应了每个AISNP的等位基因频率在不同人群间的差异程度:

H为总群体的杂合度(total heterozygosity);H为亚群体的平均杂合度(subpopulations heterozygosity)。
为人群,取值=1–;为等位基因,取值= 1–。SNP为双等位基因,故=1–2。
此外,变量之间的高度相关性,即多重共线性,会严重干扰机器学习模型训练的精准度,导致系数估计的标准误急剧增加[44,45]。因此,使用SPSS v26共线性诊断[46]筛选SNP组合,可以改进多元变量共线性问题。
与此同时,MDA交叉验证是随机森林[47]模型筛选SNP的方案,计算每个SNP的MDA值,并从大到小排序,将SNP逐一纳入模型,通过参考集十折交叉验证计算模型误差变化曲线,选择误差值最低点(±5)的SNP组合[27](图1)。
1.8 模型的选择
机器学习算法应用于解决目标对象的预测和分类问题[48,49],通常分为监督学习和无监督学习等。目前常用的监督学习类型[50]主要包括线性回归[51]、逻辑回归[52]、支持向量机(support vector machine, SVM)[53]、决策树[54]、随机森林[47]和Adaboost[55]算法等。线性回归是用来确定两种或两种以上变量间相互依赖的定量关系的一种统计方法,如最小二乘法[56]。逻辑回归是一种广义的线性回归分析模型,借助Sigmoid函数将输入的变量映射到(0~1)的区间,解决二分类问题。而softmax函数[46]是Sigmoid函数在多分类问题上的推广,将多维的输入变量映射成多维向量,而每个向量元素都在(0~1)之间。函数softmax是非常常用的逻辑回归模型,建模速度较快,尤其是解决多分类问题,易实现且计算量小、速度快。SVM主要用于解决小样本的二分类和回归问题,其基本模型定义为特征空间上的间隔最大的线性分类器,寻找一个满足分类要求的最优分类超平面,使得该超平面在保证分类精度的同时,能够使超平面两侧的空白区域最大化。但SVM算法也存在一些问题,包括训练算法速度慢、算法复杂而难以实现、测试阶段运算量大、抗击噪声及孤立点能力差等。
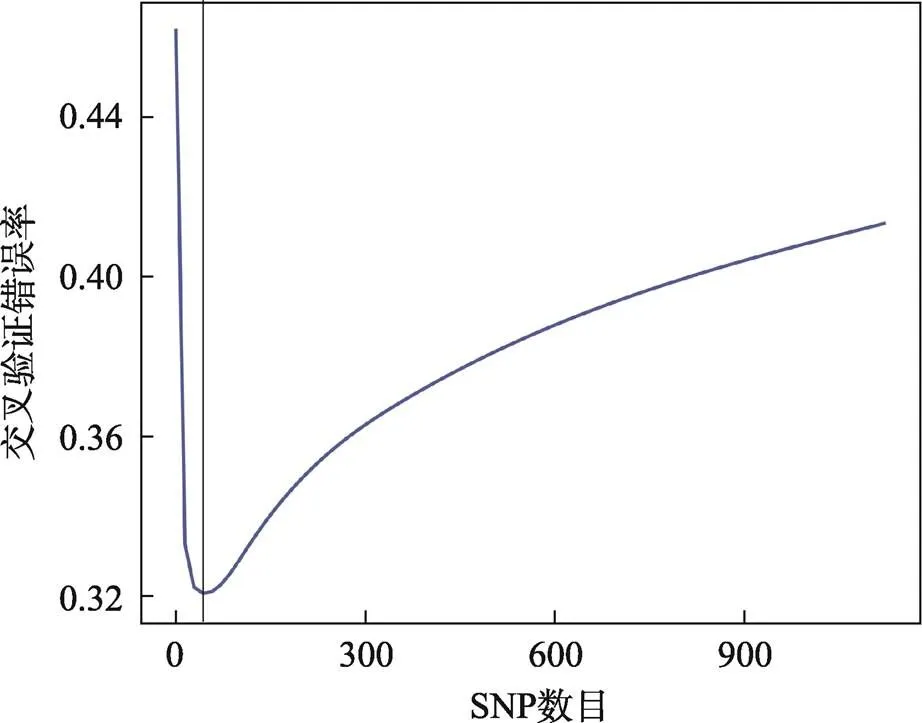
图1 AISNP的交叉验证错误率
决策树算法是一大类典型的分类方法,通过一系列规则对数据进行分类,但容易忽略变量间的相互关联并发生过拟合。而基于决策树衍生出包括随机森林、Adaboost、gradient boosting算法等。随机森林是由很多无关联的决策树构成的,能反馈高维度数据中的重要特征值,以及可以平衡误差和纠正决策树的过度拟合问题。算法Adaboost是通过训练同一个训练集不同的分类器(弱分类器),然后把这些弱分类器集合起来,构成一个更强的最终分类器(强分类器)。虽然Adaboost算法具有很高的精度,但是弱分类器数目不太好设定并且训练比较耗时,从而导致分类精度下降。Gradient boosting (如GBTD、XGboost)是一类基于梯度boosting的集成学习算法,其原理是通过弱分类器的迭代计算实现准确的分类效果,多棵决策树的所有结论累加起来作为最终的预测结果,可快速运行大规模数据,而计算相对耗时。
最后,考虑到本研究是构建人群多分类模型,样本量与数据规模较小,因此在上述机器学习算法中分别选择了最常用且适合小规模数据收敛的softmax回归算法和决策树方案的随机森林算法。
1.9 模型构建与评估
为了确保模型的稳定性,在构建模型时设置了随机数(set.seed)[57],并使用五次十折交叉验证方法[58](reateDataPartition函数)。通过将数据集样本分成10份,轮流将其中9份作为训练数据,1份作为测试数据,进行试验,从而提升模型的准确性。
softmax模型:利用R v4.0.2中的softmaxreg1.2的createDataPartition函数将参考集样本按9:1分为训练集和测试集,根据trainModel函数并通过设置隐藏节点(hidden)和选择适合的算法(algorithm)等构建预测模型。最后根据预测结果中的准确性和kappa系数选择合适的AISNP组合。其中,SNP输入数量为,隐藏层为+1,最大迭代次数Maxit为50。
随机森林模型:是先根据R4.0.2中randomForest4.6-14的randomForest函数计算每个SNP的MDA (平均精度),按MDA值从大到小排序从而确定SNP的显著性。然后根据MDA值和十折交叉验证方法(createDataPartition函数和replicate函数)得到SNP数与交叉验证错误率之间的曲线图(ggplot2包)。最后使用交叉错误率最低的SNPs构建预测模型(randomForest函数),同样根据预测结果中的准确性和kappa系数选择合适的AISNP组合。
最终,将所有挑选出的若干AISNP组合依次用softmax和随机森林模型进行评估,通过比较准确性、kappa系数、灵敏度和特异性等选择目标人群区分效果最佳的AISNP组合与机器学习模型。同时,使用DAA软件[59]进行主成分分析(principal component analysis, PCA)[60]和STRUCTURE[61]分析,基于SNP组合对目标人群的聚类和祖先成分进行比较评估。
2 结果与分析
2.1 234-AISNP组合的筛选与模型评估
通过频率差异分析技术、共线性诊断和MDA交叉验证方法并以不同阈值的和r进行AISNP的过滤和筛选,本研究得到了18组AISNPs组合。首先将这些AISNPs进行合并去重,然后按0至1128的数目进行梯度划分,即通过R4.0.2的sample函数以100个位点间隔分别在0~100、101~200、…、1001~1128这11个梯度中随机选择。最后,使用这11个AISNPs组合在参考集中的基因分型数据来构建softmax和随机森林模型,并利用对应AISNPs的测试集基因分型数据进行南-北方汉族人、韩国人和日本人的分类预测。两个模型的准确率及其95%的置信区间、kappa系数等评价指数见表4。
通过观察分析发现,两个模型的准确率随着AISNP的增加呈非线性变化,但随机森林模型的准确率变化较为平稳。其中,234-AISNP组合在softmax模型中表现最优、准确率为91.96%,735-AISNP组合在随机森林模型中表现最优、准确率为94.47%。
同时,为了评估这11个AISNP组合在目标人群中的区分效果,利用 DAA软件进行STRUCTURE和PCA分析(图2,图3)。结果发现,STRUCTURE结果K=4时,南-北方汉族人、韩国人和日本人的区分效果随着SNP数的增加呈现清晰、模糊、清晰、模糊的变化。PCA结果显示,随着SNP数减少,4个人群的区分效果表现出混乱、分散、聚集的趋势。234-AISNP组合和735-AISNP组合在这两个方案中均达到了目标人群高度区分的效果。

表4 在softmax和随机森林模型中参考集与测试集的表现评估
红色字体为两组最优位点组合。
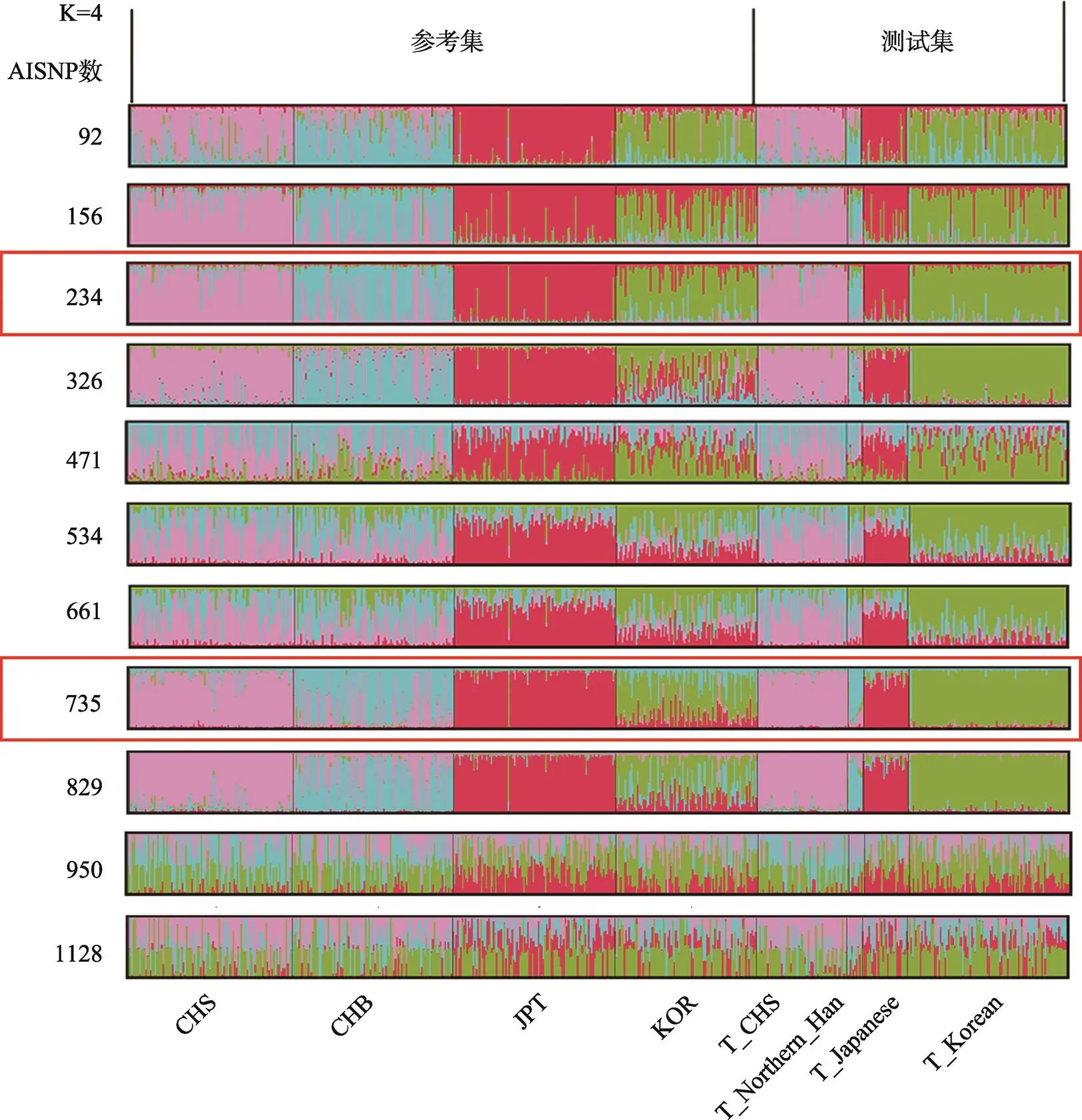
图2 11种AISNP组合的祖先成分分析结果
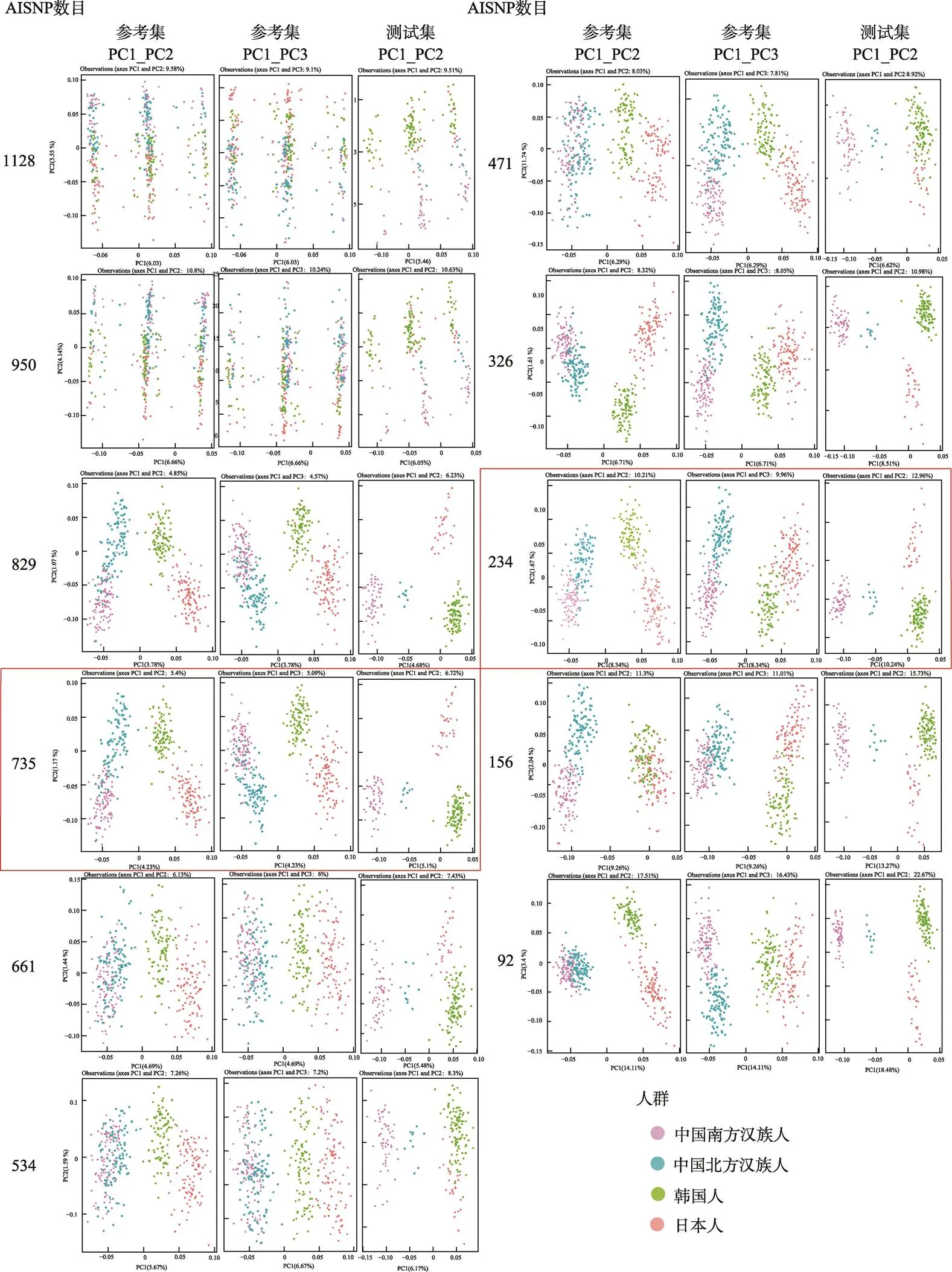
图3 11种AISNP组合的PCA分析图
最后,综合比较234-AISNP组合和735-AISNP组合在两种机器学习模型(softmax和随机森林)、STRUCTURE和PCA分析中南-北方汉族人、韩国人和日本人的区分效果。结果表明,735-AISNP组合在随机森林模型、STRUCTURE和PCA分析中均实现了4个人群的遗传划分,但是在softmax模型中没有达到预期区分效果。尽管234-AISNP组合在随机森林模型中的准确率低于735-AISNP组合的准确率,但234-AISNP组合在softmax模型中的准确率是最高的,并且该组合在STRUCTURE和PCA分析中也实现了目标人群的遗传划分,故234-AISNP组合的区分效果最好(表5)。

表5 234-AISNP组合的信息
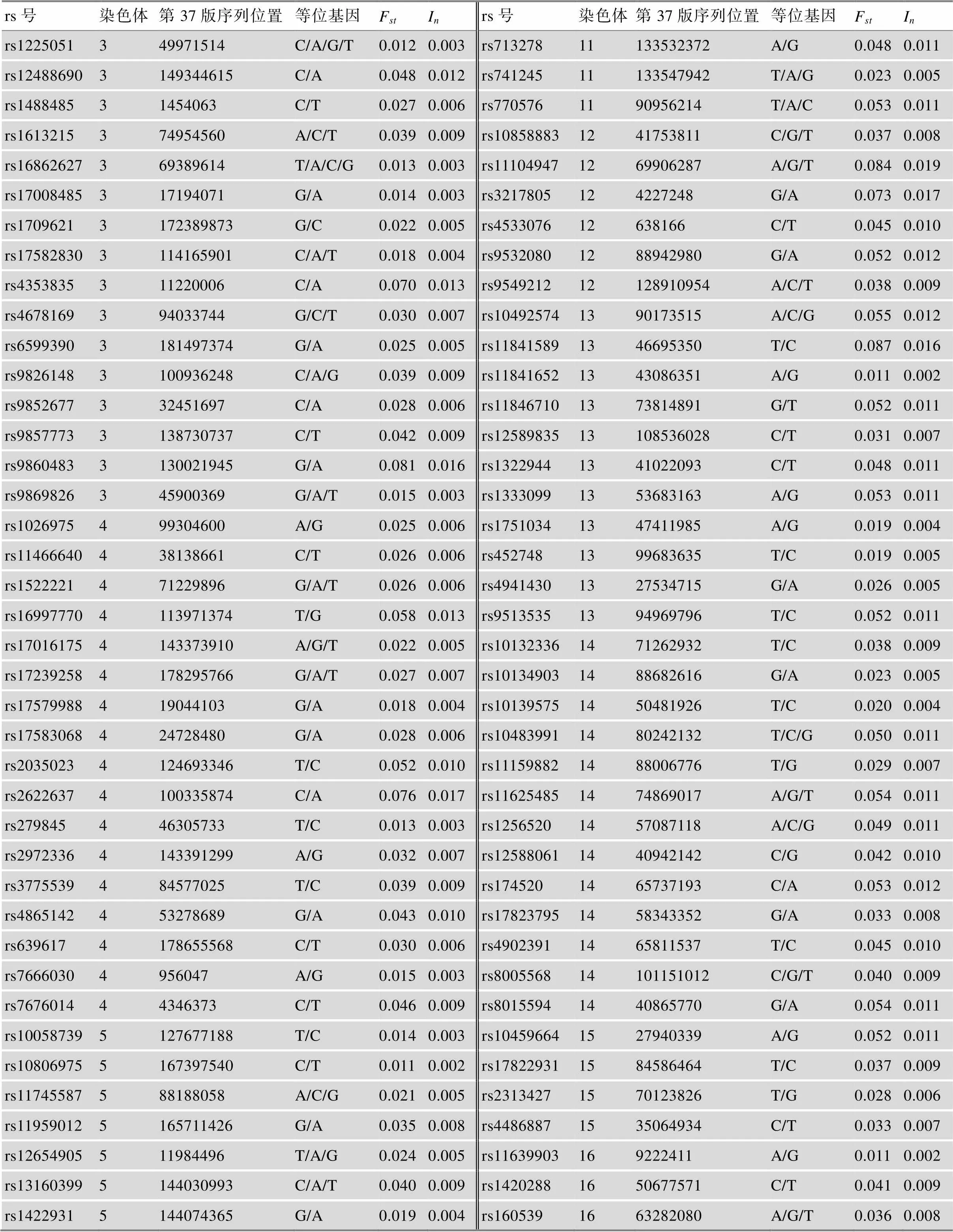
续表
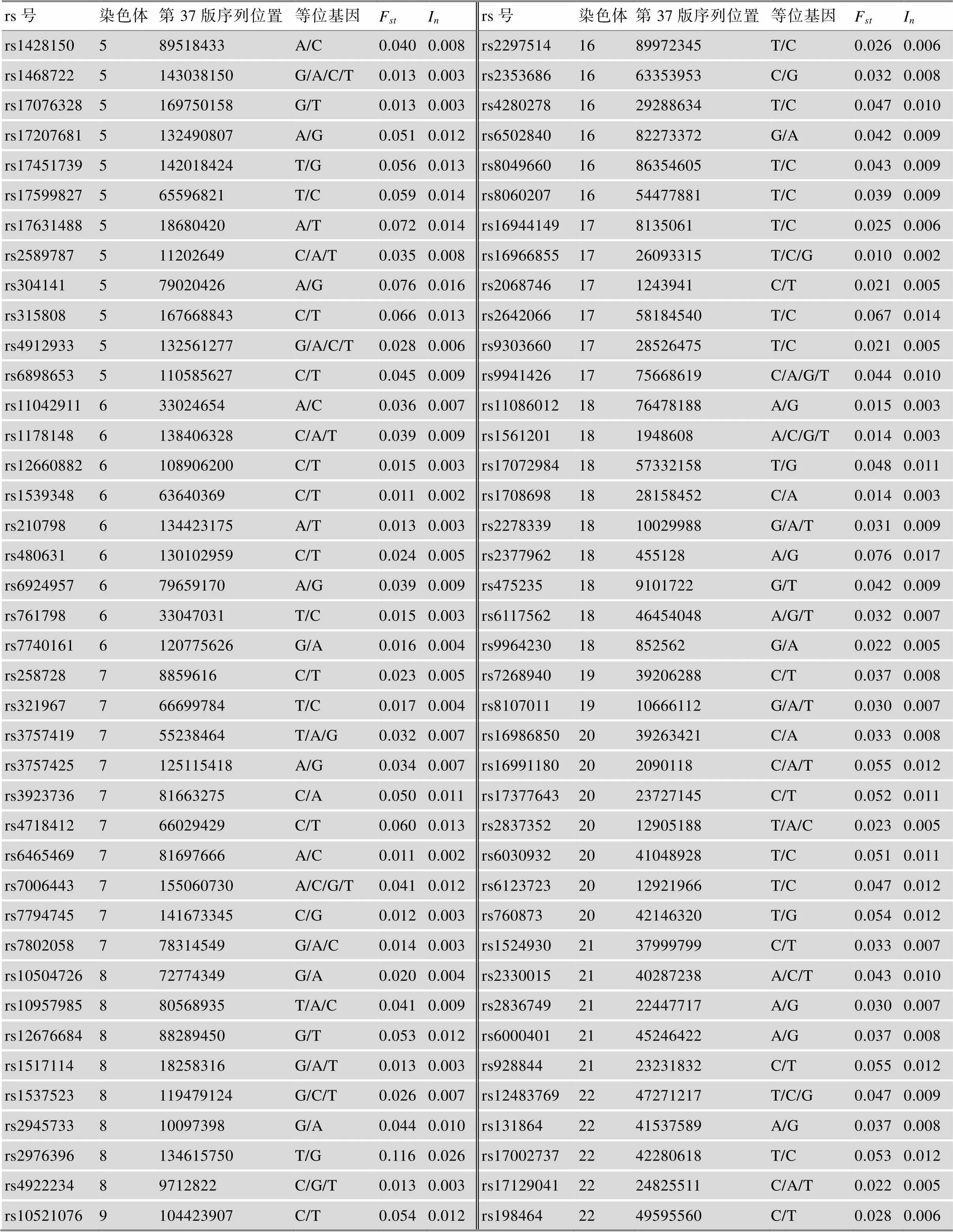
续表
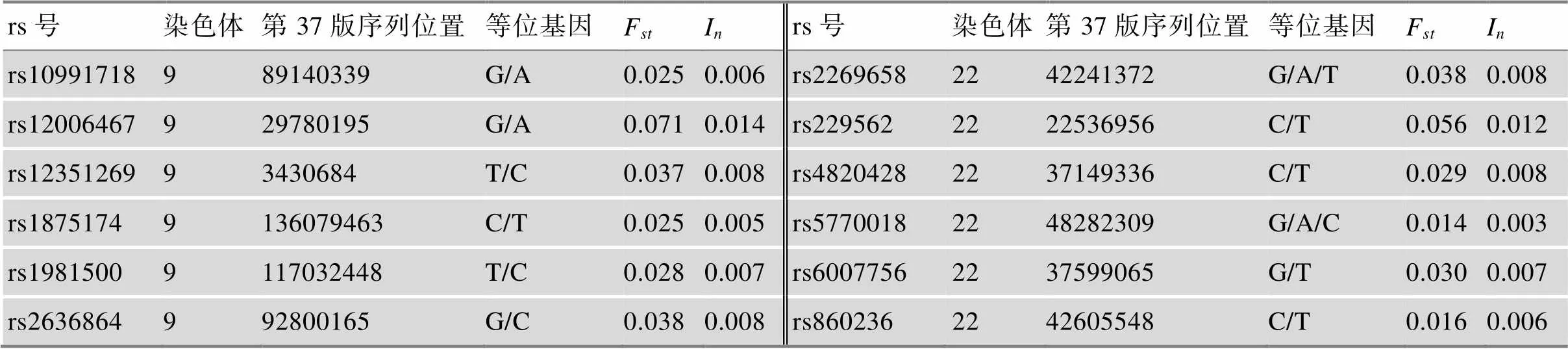
续表
2.2 234-AISNP组合的人群聚类效果提升
为了进一步评估234-AISNP组合在区分南-北方汉族人、韩国人和日本人的效果,基于1128和234个AISNP的基因分型,分别对403份参考集和199份测试集样本进行系统发育树构建(图4,A和B),结果显示4个人群呈现一定区分度。
1128-AISNP组合的参考集结果表明(图4A),南北方汉族人存在部分个体混合,韩国人呈两簇分布,与日本人和汉族人相邻。测试集结果显示,北方汉族人与南方汉族人、日本人交叉混合,少部分韩国人和日本人聚类。
234-AISNP组合的参考集结果表明(图4B),南-北方汉族人混合的个体数明显减少,仅少数的韩国人与北方汉族人聚类,且日本人与韩国人聚类,无个体的混合。测试集结果显示,北方汉族人的聚类效果得到提升,与少部分南方汉族人和日本人混合,虽然部分韩国人与日本人聚为一簇,但无混合。从整体来看,相比1128-AISNP组合,234-AISNP组合的聚类分析结果更优。
3 讨论
法医DNA鉴定技术作为打击犯罪的核心技术手段之一,为维护社会的治安稳定发挥着关键性作用[62]。SNP族群推断技术[63]作为对现有DNA比对技术的有力补充,通过更深层次解读生物物证的遗传信息,对DNA来源人的种族地域来源和外形体貌特征进行遗传推断和刻画,从而最大程度的发挥“生物证人”的作用,为案件侦破提供全新的线索,对判断嫌疑人的种族来源、定义案件的性质起到了至关重要的作用[63]。
本研究通过频率差异分析技术、共线性诊断和MDA交叉验证方法,并结合HWE、LD筛选出了不同SNP组合,以测试两种机器学习模型(逻辑回归算法softmax模型和决策树算法随机森林模型)、STRUCTURE (贝叶斯聚类)、PCA (协方差)和系统发育树(皮尔逊相关系数)方法对南-北汉族、韩国和日本人群的分类效果。这些SNP组合在这些方案中都将4个人群不同程度的区分开,并且这4种方法对人群的区分或聚类效果是不同的。例如SNP数为234~534时,softmax模型准确率均大于90%,但STRUCTURE和PCA分析的结果除234-AISNP组合外,其他SNP组合未将4个人群精确区分开。另外,735和829 AISNPs在随机森林模型中准确率达到90%,并且STRUCTURE和PCA分析结果实现了目标人群的遗传划分,但是这两组SNP在softmax模型中的准确率仅为14.57%和44.22%。因此,只有选择恰当的SNP组合与判别方法才能达到最佳的人群区分效果。本研究最终挑选的234-AISNP组合在4个方案中都达到了南-北方汉族人、韩国人和日本人精确区分的目的,且SNP数较少,适合法医学应用。由于本研究中仅使用了千人基因组数据库中的汉族样本作为机器学习的参考数据集,有限的样本量限制或掩盖了某些AISNP真正的识别能力,后续将继续增加样本量与数据来源,进一步验证和优化234个AISNP组合。
在模型评估分析中,本研究发现softmax模型的准确率随着SNP数目增加,呈现出上升、到达最高峰(91.96%)后稳定、再下降(14.57%)、最后上升(48.24%)的趋势(表4),而随机森林模型的准确率变化较稳定(平均83.46%)。未经筛选的AISNP位点中存在一定比例的共线性问题即自变量间存在较强的相关性,而其中部分位点的低差异性干扰了差异信息的提炼,因此表现出高度的群体相似性。本研究中,过多AISNP的输入严重干扰机器学习模型训练和测试的精准度,并出现模型出现过拟合[64]以适应训练数据,从而在测试数据上效果很差。使用共线性诊断和MDA交叉验证对AISNP进行过滤,以提升模型的准确率。同时,过少AISNP的输入会导致模型出现欠拟合现象[65],使得模型在训练和测试数据集上的效果都很差。综上所述,在众多特征中需要将无关和冗余特征去除,使合适的特征纳入模型,才能够进一步提升模型的稳定性和准确率。同时本研究也观察到,构建的两个模型在目标人群区分时,更集中于韩国人和中国南方汉族人的区分,忽略了中国北方汉族人的区分,最终导致模型测试准确率较低并且相应的评价参数值也较低。这些可能是由于参考集样本少导致机器学习模型不稳定,或者是测试集样本数目的不均衡导致模型出现欠拟合以及模型对目标人群的不平衡区分,从而使得模型测试结果不佳。为了解决这些问题,本研究在构建目标人群的预测和测试模型时,不仅设置了随机数和调试最大迭代次数,还使用了五次十折交叉验证方法,从而达到提升模型准确率的目的,最终实现目标人群的精确区分。另外,还观察到SNP数增加对softmax模型系统性能的影响更加明显,并且当SNP数目增加到某个阈值时,两个模型均达到饱和即准确率不存在过大波动。
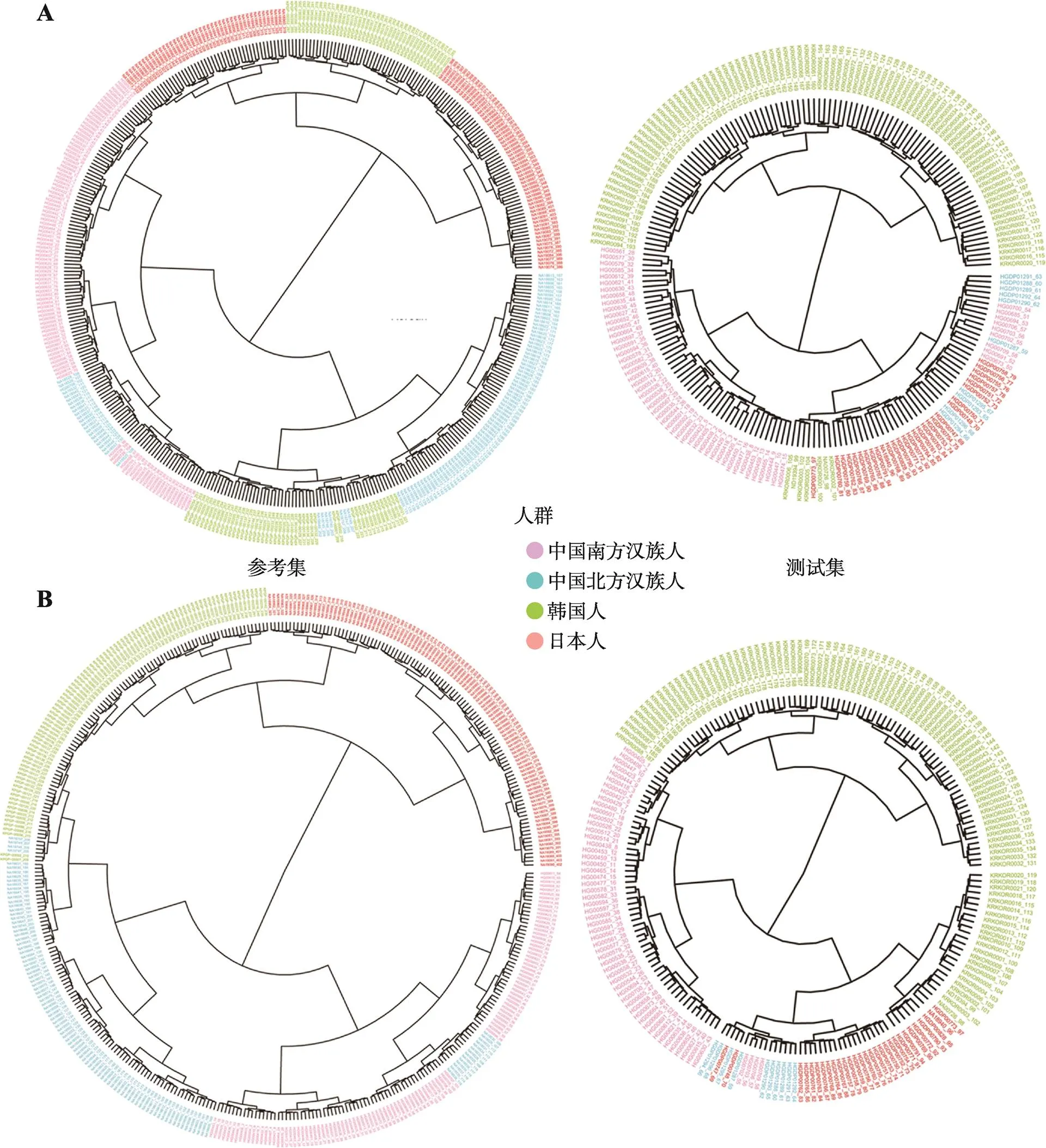
图4 基于1128-AISNP组合与234-AISNP组合的基因分型绘制的目标人群的系统发育树
A:1128-AISNP组合的参考及测试集中目标人群的系统发育树结果;B:234-AISNP组合的参考及测试集中目标人群的系统发育树结果。
总之,本研究先利用频率差异分析技术(F、I、HWE和LD)及两种机器学习算法(softmax和随机森林),筛选AISNP、建立目标人群遗传推断模型并测试,再结合经典族群推断算法(STRUCTURE和PCA)[59]对AISNP组合进行评估,最终234-AISNP组合在这些方法中均实现了南-北方汉族人、韩国人和日本人的精确区分。其次,本研究发现softmax模型和MDA交叉验证运行速度过慢,需要收集更加高效快速的筛选AISNP方法和更精准构建人群遗传划分模型的机器学习方法。最后,在参考集和测试集样本收集的时候,需要考虑其来源是否多样、数目是否庞大、以及检测方法是否相同等问题。综上所述,在进行AISNP筛选和近距离目标群体区分时,尽可能增加参考集和测试集样本量并运用不同方法多方面综合评估,从而选出高效能高质量的AISNP组合,为法医学基础数据库进行扩充。
感谢中国科学院上海生命科学研究院计算生物学研究所的徐书华老师在文章数据方面给予的帮助。
附加材料见文章电子版www.chinagene.cn。
[1] Phillips C. Forensic genetic analysis of bio-geographical ancestry., 2015, 18: 49–65.
[2] Tishkoff SA, Kidd KK. Implications of biogeography of human populations for 'race' and medicine., 2004, 36(11 Suppl): S21–S27.
[3] Marchini J, Cardon LR, Phillips MS, Donnelly P. The effects of human population structure on large genetic association studies., 2004, 36(5): 512–517.
[4] Paschou P, Lewis J, Javed A, Drineas P. Ancestry informative markers for fine-scale individual assignment to worldwide populations., 2010, 47(12): 835–847.
[5] Phillips C, Salas A, Sánchez JJ, Fondevila M, Gómez-Tato A, Alvarez-Dios J, Calaza M, de Cal MC, Ballard D, Lareu MV, Carracedo A. Inferring ancestral origin using a single multiplex assay of ancestry-informative marker SNPs., 2007, 1(3–4): 273–280.
[6] Kidd KK, Speed WC, Pakstis AJ, Furtado MR, Fang RX, Madbouly A, Maiers M, Middha M, Friedlaender FR, Kidd JR. Progress toward an efficient panel of SNPs for ancestry inference., 2014, 10: 23–32.
[7] Jiang L, Sun QF, Ma Q, Zhao WT, Liu J, Zhao L, Ji AQ, Li CX. Optimization and validation of analysis method based on 27-plex SNP panel for ancestry inference., 2017, 39(2): 166–173.
江丽, 孙启凡, 马泉, 赵雯婷, 刘京, 赵蕾, 季安全, 李彩霞. 27-plex SNP 种族推断方法的优化及验证. 遗传, 2017, 39(2): 166–173.
[8] Qin PF, Li ZQ, Jin WF, Lu DS, Lou HY, Shen JW, Jin L, Shi YY, Xu SH. A panel of ancestry informative markers to estimate and correct potential effects of population stratification in Han Chinese., 2014, 22(2): 248–253.
[9] Wang YC, Lu DS, Chung YJ, Xu SH. Genetic structure, divergence and admixture of Han Chinese, Japanese and Korean populations., 2018, 155: 19.
[10] Shi CM, Liu Q, Zhao SL, Chen H. Ancestry informative SNP panels for discriminating the major East Asian populations: Han Chinese, Japanese and Korean., 2019, 83(5): 348–354.
[11] Wang CC, Yeh HY, Popov AN, Zhang HQ, Matsumura H, Sirak K, Cheronet O, Kovalev A, Rohland N, Kim AM, Mallick S, Bernardos R, Tumen D, Zhao J, Liu YC, Liu JY, Mah M, Wang K, Zhang Z, Adamski N, Broomandkhoshbacht N, Callan K, Candilio F, Carlson KSD, Culleton BJ, Eccles L, Freilich S, Keating D, Lawson AM, Mandl K, Michel M, Oppenheimer J, Özdoğan KT, Stewardson K, Wen SQ, Yan S, Zalzala F, Chuang R, Huang CJ, Looh H, Shiung CC, Nikitin YG, Tabarev AV, Tishkin AA, Lin S, Sun ZY, Wu XM, Yang TL, Hu X, Chen L, Du H, Bayarsaikhan J, Mijiddorj E, Erdenebaatar D, Iderkhangai TO, Myagmar E, Kanzawa-Kiriyama H, Nishino M, Shinoda KI, Shubina OA, Guo J, Cai WW, Deng QY, Kang LL, Li D, Li DW, Lin RM, Nini, Shrestha R, Wang LX, Wei LW, Xie GM, Yao HB, Zhang MF, He GL, Yang XM, Hu R, Robbeets M, Schiffels S, Kennett DJ, Jin L, Li H, Krause J, Pinhasi R, Reich D. Genomic insights into the formation of human populations in East Asia., 2021, 591(7850): 413–419.
[12] Jung JY, Kang PW, Kim E, Chacon D, Beck D, McNevin D. Ancestry informative markers (AIMs) for Korean and other East Asian and South East Asian populations., 2019, 133(6): 1711–1719.
[13] Okada Y, Momozawa Y, Sakaue S, Kanai M, Ishigaki K, Akiyama M, Kishikawa T, Arai Y, Sasaki T, Kosaki K, Suematsu M, Matsuda K, Yamamoto K, Kubo M, Hirose N, Kamatani Y. Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese., 2018, 9(1): 1631.
[14] Akiyama M, Okada Y, Kanai M, Takahashi A, Momozawa Y, Ikeda M, Iwata N, Ikegawa S, Hirata M, Matsuda K, Iwasaki M, Yamaji T, Sawada N, Hachiya T, Tanno K, Shimizu A, Hozawa A, Minegishi N, Tsugane S, Yamamoto M, Kubo M, Kamatani Y. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population., 2017, 49(10): 1458–1467.
[15] Liu SY, Huang SJ, Chen F, Zhao LJ, Yuan YY, Francis SS, Fang L, Li ZL, Lin L, Liu R, Zhang Y, Xu HX, Li SK, Zhou YW, Davies RW, Liu Q, Walters RG, Lin K, Ju J, Korneliussen T, Yang MA, Fu QM, Wang J, Zhou LJ, Krogh A, Zhang HY, Wang W, Chen ZM, Cai ZM, Yin Y, Yang HM, Mao M, Shendure J, Wang J, Albrechtsen A, Jin X, Nielsen R, Xu X. Genomic analyses from non-invasive prenatal testing reveal genetic associations, patterns of viral infections, and Chinese population history., 2018, 175(2): 347–359.
[16] Xu SH, Yin XY, Li SL, Jin WF, Lou HY, Yang L, Gong XH, Wang HY, Shen YP, Pan XD, He YG, Yang YJ, Wang Y, Fu WQ, An Y, Wang JC, Tan JZ, Qian J, Chen XL, Zhang X, Sun YF, Zhang XJ, Wu BL, Jin L. Genomic dissection of population substructure of Han Chinese and its implication in association studies., 2009, 85(6): 762–774.
[17] Jeon S, Bhak Y, Choi Y, Jeon Y, Kim S, Jang J, Jang J, Blazyte A, Kim C, Kim Y, Shim J, Kim N, Kim YJ, Park SG, Kim J, Cho YS, Park Y, Kim HM, Kim BC, Park NH, Shin ES, Kim BC, Bolser D, Manica A, Edwards JS, Church G, Lee S, Bhak J. Korean genome project: 1094 Korean personal genomes with clinical information., 2020, 6(22): eaaz7835.
[18] Cao YN, Li L, Xu M, Feng ZM, Sun XH, Lu JL, Xu Y, Du PN, Wang TG, Hu RY, Ye Z, Shi LX, Tang XL, Yan L, Gao ZN, Chen G, Zhang YF, Chen LL, Ning G, Bi YF, Wang WQ, Consortium C. The ChinaMAP analytics of deep whole genome sequences in 10,588 individuals., 2020, 30(9): 717–731.
[19] Jinam TA, Kanzawa-Kiriyama H, Inoue I, Tokunaga K, Omoto K, Saitou N. Unique characteristics of the Ainu population in Northern Japan., 2015, 60(10): 565–571.
[20] Kim JJ, Verdu P, Pakstis AJ, Speed WC, Kidd JR, Kidd KK. Use of autosomal loci for clustering individuals and populations of East Asian origin., 2005, 117(6): 511–519.
[21] Clarke L, Fairley S, Zheng-Bradley X, Streeter I, Perry E, Lowy E, Tassé A-M, and Flicek P. The international genome sample resource (IGSR): a worldwide collection of genome variation incorporating the 1000 genomes project data., 2016, 45(1): 854–859.
[22] Zhang WQ, Meehan J, Su ZQ, Ng HW, Shu M, Luo H, Ge WG, Perkins R, Tong WD, Hong HX. Whole genome sequencing of 35 individuals provides insights into the genetic architecture of Korean population., 2014, 15(11): 6–18.
[23] Byrska-Bishop M, Evani US, Zhao XF, Basile AO, Abel HJ, Regier AA, Corvelo A, Clarke WE, Musunuri R, Nagulapalli K, Fairley S, Runnels A, Winterkorn L, Lowy E, Flicek P, Germer S, Brand H, Hall IM, Talkowski ME, Narzisi G, Zody MC, The Human Genome Structural Variation Consortium. High coverage whole genome sequencing of the expanded 1000 Genomes Project cohort including 602 trios., 2021, doi: 10.1101/2021. 02.06.430068.
[24] Bergström A, McCarthy SA, Hui RY, Almarri MA, Ayub Q, Danecek P, Chen Y, Felkel S, Hallast P, Kamm J, Blanché H, Deleuze JF, Cann H, Mallick S, Reich D, Sandhu MS, Skoglund P, Scally A, Xue YL, Durbin R, Tyler-Smith C. Insights into human genetic variation and population history from 929 diverse genomes., 2020, 367(6484): eaay5012.
[25] Mallick S, Li H, Lipson M, Mathieson I, Gymrek M, Racimo F, Zhao MY, Chennagiri N, Nordenfelt S, Tandon A, Skoglund P, Lazaridis I, Sankararaman S, Fu QM, Rohland N, Renaud G, Erlich Y, Willems T, Gallo C, Spence JP, Song YS, Poletti G, Balloux F, van Driem G, de Knijff P, Romero IG, Jha AR, Behar DM, Bravi CM, Capelli C, Hervig T, Moreno-Estrada A, Posukh OL, Balanovska E, Balanovsky O, Karachanak-Yankova S, Sahakyan H, Toncheva D, Yepiskoposyan L, Tyler-Smith C, Xue YL, Abdullah MS, Ruiz-Linares A, Beall CM, Di Rienzo A, Jeong C, Starikovskaya EB, Metspalu E, Parik J, Villems R, Henn BM, Hodoglugil U, Mahley R, Sajantila A, Stamatoyannopoulos G, Wee JTS, Khusainova R, Khusnutdinova E, Litvinov S, Ayodo G, Comas D, Hammer MF, Kivisild T, Klitz W, Winkler CA, Labuda D, Bamshad M, Jorde LB, Tishkoff SA, Watkins WS, Metspalu M, Dryomov S, Sukernik R, Singh L, Thangaraj K, Pääbo S, Kelso J, Patterson N, Reich D. The Simons genome diversity project: 300 genomes from 142 diverse populations., 2016, 538(7624): 201–206.
[26] Liu XY, Lu DS, Saw WY, Shaw PJ, Wangkumhang P, Ngamphiw C, Fucharoen S, Lert-Itthiporn W, Chin- Inmanu K, Chau TNB, Anders K, Kasturiratne A, de Silva HJ, Katsuya T, Kimura R, Nabika T, Ohkubo T, Tabara Y, Takeuchi F, Yamamoto K, Yokota M, Mamatyusupu D, Yang WJ, Chung YJ, Jin L, Hoh BP, Wickremasinghe AR, Ong RH, Khor CC, Dunstan SJ, Simmons C, Tongsima S, Suriyaphol P, Kato N, Xu SH, Teo YY. Characterising private and shared signatures of positive selection in 37 Asian populations., 2017, 25(4): 499–508.
[27] Wen H, Wei YL, Guo XY, Sun CC, Xue SY, Liu J, Fan H, Jiang L. High-resolution SNP ancestry inference model and efficiency evaluation in three East Asian populations., 2021, 48(8): 973–981.
文豪, 魏以梁, 郭晓媛, 孙昌春, 薛思瑶, 刘京, 范虹, 江丽. 东亚三族群SNP高分辨推断模型构建与效能评估. 生物化学与生物物理进展, 2021, 48(8): 973–981.
[28] Guo XY, Sun CC, Xue SY, Zhao H, Jiang L, Li CX. 49AISNP: a study on the ancestry inference of the three ethnic groups in the north of East Asia., 2021, 43(9): 880–889.
郭晓媛, 孙昌春, 薛思瑶, 赵慧, 江丽, 李彩霞. 49AISNP:东亚北方三个族群遗传来源推断. 遗传, 2021, 43(9): 880–889.
[29] Kim T, Seo HD, Hennighausen L, Lee D, Kang K. Octopus-toolkit: a workflow to automate mining of public epigenomic and transcriptomic next-generation sequencing data., 2018, 46(9): 53–58.
[30] 1000 Genomes Project Consortium, Auton A, Brooks LD, Durbin RM, Garrison EP, Kang HM, Korbel JO, Marchini JL, McCarthy S, McVean GA, Abecasis GR. A global reference for human genetic variation., 2015, 526(7571): 68–74.
[31] Sudmant PH, Rausch T, Gardner EJ, Handsaker RE, Abyzov A, Huddleston J, Zhang Y, Ye K, Jun G, Fritz MHY, Konkel MK, Malhotra A, Stütz AM, Shi XH, Casale FP, Chen JM, Hormozdiari F, Dayama G, Chen K, Malig M, Chaisson MJP, Walter K, Meiers S, Kashin S, Garrison E, Auton A, Lam HYK, Mu XJ, Alkan C, Antaki D, Bae T, Cerveira E, Chines P, Chong ZC, Clarke L, Dal E, Ding L, Emery S, Fan X, Gujral M, Kahveci F, Kidd JM, Kong Y, Lameijer EW, McCarthy S, Flicek P, Gibbs RA, Marth G, Mason CE, Menelaou A, Muzny DM, Nelson BJ, Noor A, Parrish NF, Pendleton M, Quitadamo A, Raeder B, Schadt EE, Romanovitch M, Schlattl A, Sebra R, Shabalin AA, Untergasser A, Walker JA, Wang M, Yu FL, Zhang C, Zhang J, Zheng-Bradley XQ, Zhou WD, Zichner T, Sebat J, Batzer MA, McCarroll SA, 1000 Genomes Project Consortium, Mills RE, Gerstein MB, Bashir A, Stegle O, Devine SE, Lee C, Eichler EE, Korbel JO. An integrated map of structural variation in 2,504 human genomes., 2015, 526(7571): 75–81.
[32] Korn JM, Kuruvilla FG, McCarroll SA, Wysoker A, Nemesh J, Cawley S, Hubbell E, Veitch J, Collins PJ, Darvishi K, Lee C, Nizzari MM, Gabriel SB, Purcell S, Daly MJ, Altshuler D. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs., 2008, 40(10): 1253–1260.
[33] Van der Auwera GA, O'Connor BD. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra. 2020: O'Reilly Media, Incorporated.
[34] Meire M, Ballings M, Van den Poel D. imputeMissings: impute missing values in a predictive context. 2016.
[35] Rustowicz R. Crop classification with multi-temporal satellite imagery. 2017.
[36] Breiman L, Cutler A, Liaw A, Wiener M. Package ‘randomForest’. 2018.
[37] Yu GC, Smith DK, Zhu HC, Guan Y, Lam TTY. ggtree: an R package for visualization and annotation of phylogenetic trees with their covariates and other associated data., 2017, 8(1): 28–36.
[38] Hao W, Storey JD. Extending tests of Hardy-Weinberg equilibrium to structured populations., 2019, 213(3): 759–770.
[39] Pritchard JK, Przeworski M. Linkage disequilibrium in humans: models and data., 2001, 69(1): 1–14.
[40] Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps., 2005, 21(2): 263–265.
[41] Armstrong RA. When to use the Bonferroni correction., 2014, 34(5): 502–508.
[42] Boca SM, Rosenberg NA. Mathematical properties of Fst between admixed populations and their parental source populations., 2011, 80(3): 208–216.
[43] Rosenberg NA, Li LM, Ward R, Pritchard JK. Informativeness of genetic markers for inference of ancestry., 2003, 73(6): 1402–1422.
[44] Hui SB, Wang WJ. Improvement of multi-variable's redundant attributes in classification algorithm of support vector machines., 2006, 27(8): 1385–138.
惠守博, 王文杰. 支持向量机分类算法中多元变量共线性问题的改进. 计算机工程与设计, 2006, 27(8): 1385– 1388.
[45] Zhao YD, Liu R, Liu YL, Xiao F, Zhang Y. Multivariate logistic regression collinearity diagnosis analysis., 2000, (5): 3–5.
赵宇东, 刘嵘, 刘延龄, 肖峰, 张扬. 多元logistic回归的共线性分析. 中国卫生统计, 2000, (5): 3–5.
[46] Wang L, Tong X, Sheng MW, Qin HD, Tang QS. Review of image classification based on softmax classifier in deep learning., 2019, 18(6): 1-9+47.
万磊, 佟鑫, 盛明伟, 秦洪德, 唐松奇. Softmax分类器深度学习图像分类方法应用综述. 导航与控制, 2019, 18(6): 1-9+47.
[47] Rigatti SJ. Random Forest., 2017, 47(1): 31–39.
[48] Heo J, Yoon JG, Park H, Kim YD, Nam HS, Heo JH. Machine learning-based model for prediction of outcomes in acute stroke., 2019, 50(5): 1263–1265.
[49] Che DS, Liu Q, Rasheed K, Tao XP. Decision tree and ensemble learning algorithms with their applications in bioinformatics., 2011, 696: 191–199.
[50] Connor CW. Artificial intelligence and machine learning in anesthesiology., 2019, 131(6): 1346– 1359.
[51] Pandis N. Linear regression., 2016, 149(3): 431–434.
[52] LaValley MP. Logistic regression., 2008, 117(18): 2395–2399.
[53] Huang SJ, Cai NG, Pacheco PP, Narrandes S, Wang Y, Xu W. Applications of support vector machine (SVM) learning in cancer genomics., 2018, 15(1): 41–51.
[54] Karalis G. Decision trees and applications., 2020, 1194: 239–242.
[55] Hatwell J, Gaber MM, Atif Azad RM. Ada-WHIPS: explaining AdaBoost classification with applications in the health sciences., 2020, 20(1): 250.
[56] Wen J, Xu Y, Li ZY, Ma ZL, Xu YR. Inter-class sparsity based discriminative least square regression., 2018, 102: 36–47.
[57] Kloumann IM, Ugander J, Kleinberg J. Block models and personalized PageRank., 2017, 114(1): 33–38.
[58] Jung Y, Hu JH. A k-fold averaging cross-validation procedure., 2015, 27(2): 167–179.
[59] Liu J, Li S, Jiang L, Zhao L, Zhao WT, Feng L, Liu HB, Ji AQ, Li CX. DNA ancestry analyzer: an automatic program for ancestry inference of unknown individuals., 2018, 22(1): 3-7+41.
刘京, 李盛, 江丽, 赵蕾, 赵雯婷, 丰蕾, 刘海渤, 季安全, 李彩霞. 对于未知来源个体进行族群推断的自动分析系统. 生命科学研究, 2018, 22(1): 3-7+41.
[60] Ringnér M. What is principal component analysis?, 2008, 26(3): 303–304.
[61] Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data., 2000, 155(2): 945–959.
[62] Cai LJ. The technical means of forensic material evidence identification in criminal investigation cases-DNA identification technology., 2018, (34): 177.
蔡立君. 刑侦案件中法医物证鉴定的技术手段——DNA鉴定技术. 法制博览, 2018, (34): 177.
[63] Jiang L, Zhao L, Liu J, Zhao WT, Ma Q, Zhao H, Ji AQ, Li CX. DNA ancestry inference assisting to have a case solved., 2019, 44(4): 371–373.
江丽, 赵蕾, 刘京, 赵雯婷, 马泉, 赵慧, 季安全, 李彩霞. DNA供者族群推断技术在案件中的应用. 刑事技术, 2019, 44(4): 371–373.
[64] Charilaou P, Battat R. Machine learning models and over-fitting considerations., 2022, 28(5): 605–607.
[65] Dizaji KG, Chen W, Huang H. Deep large-scale multitask learning network for gene expression inference., 2021, 28(5): 485–500.
Optimization scheme of machine learning model for genetic division between northern Han, southern Han, Korean and Japanese
Yongqiang Kong1, Jinkai Liu1, Jiaqi Gu2, Jingyi Xu1, Yunuo Zheng2, Yiliang Wei2, Shaoyuan Wu1,2
Han Chinese, Korean and Japanese are the main populations of East Asia, and Han Chinese presents a gradient admixture from north to south. There are differences among the East Asian populations in genetic structure. To achieve fine-scale genetic classification of southern (S-) and northern (N-) Han Chinese, Korean and Japanese individuals in this study, we collected and analyzed 1185 ancestry informative SNPs (AISNPs) from previous literature reports and our laboratory findings. First, two machine learning algorithms, softmax and randomForest, were used to build genetic classification models. Then, phylogenetic tree, STRUCTURE and principal component analysis were used to evaluate the performance of classification for different AISNP panels. The 234-AISNP panel achieved a fine-scale differentiation among the target populations in four classification schemes. The accuracy of the softmax model was 92%, which realized the accurate classification of the S-Han, N-Han, Korean and Japanese individuals. The two machine learning models tested in this study provided important references for the high-resolution discrimination of close-range populations and will be useful tools to optimize marker panels for developing forensic DNA ancestry inference systems.
forensic genetics; ancestry informative SNPs; machine learning; East Asia; S-Han and N-Han
2022-05-03;
2022-07-13;
2022-08-11
法医遗传学公安部重点实验室开放课题(编号:2020FGKFKT01),江苏省研究生科研与实践创新计划项目任务书(编号:KYCX20_2286,KYCX21_2597)资助[Supported by the Key Laboratory of Forensic Genetics of China (No. 2020FGKFKT01), the Graduate Research and Practice Innovation Program of Jiangsu Normal University (Nos. KYCX20_2286,KYCX21_2597)]
孔永强,在读硕士研究生,专业方向:生物学。E-mail: kongyongqiang@tmu.edu.cn
魏以梁,博士,副教授,研究方向:法医遗传学。E-mail: weiyiliang.2013@tsinghua.org.cn
伍少远,博士,教授,研究方向:系统发育与比较基因组学。E-mail: shaoyuan5@gmail.com
10.16288/j.yczz.22-073
(责任编委: 朱波峰)
猜你喜欢
区域治理(2022年40期)2022-11-27
小学生学习指导(高年级)(2022年10期)2022-11-04
健康之家(2021年19期)2021-05-23
医学食疗与健康(2021年27期)2021-05-13
农业科技与信息(2021年2期)2021-03-27
健康体检与管理(2021年10期)2021-01-03
动漫界·幼教365(小班)(2019年10期)2019-10-28
动漫界·幼教365(大班)(2019年10期)2019-10-28
动漫界·幼教365(中班)(2019年10期)2019-10-28
奥秘(创新大赛)(2019年3期)2019-03-13
