
17α-羟化酶/17,20-裂解酶缺陷症致先天性肾上腺皮质增生病例分析
2022-11-25 01:32文重远
微循环学杂志 2022年4期
包 艳 易 波 王 晔 文重远
先天性肾上腺皮质增生症(Congenital Adrenal Hyperplasia, CAH)为一组常染色体隐性遗传病,是由于参与肾上腺类固醇激素合成的催化酶活性受损,造成相应激素生成障碍所致[1]。其临床表现取决于发生缺陷酶的种类和受损程度。CAH包括21-羟化酶缺陷症(最常见,90%以上)、17α-羟化酶/17,20-裂解酶缺陷症(17α-hydroxylation/17,20-lyase deficiency, 17OHD)、3β-类固醇脱氢酶缺陷症、11β-羟化酶缺陷症和类固醇激素合成急性调节蛋白缺陷症等。17OHD是其中一种罕见类型,由CYP17A1基因突变所致,约占CAH的1%,全球发病率约为1∶50 000-100 000[2]。笔者通过对一家系中同患17OHD两“姐妹”的临床表现、实验室检查及基因突变类型的分析,并复习国内外相关文献,发现该病罕见,临床表现多样化,极易漏诊、误诊。故报道如下,以期提高临床医生对该病的认识。
1 资料与方法
1.1 病例资料
患者1(先证者),社会性别:女,15岁,2019年7月因“高血压”前往本院就诊。在院外测得最高血压为170/120mmHg,曾在当地医院给予厄贝沙坦治疗,但血压控制效果欠佳。家族史:其父母非近亲结婚;其姐姐有月经不调病史。既往史:曾行“腹股沟疝修补术”。查体:身高171cm,体重58kg,血压150/100mmHg。乳房Tanner Ⅰ期,无腋毛。外阴幼女型,可探及约3cm的盲端,Tanner Ⅰ期。腹股沟未触及包块。未见睾丸和阴茎。
患者2,先证者姐姐,社会性别:女,21岁,2019年间多次以“月经量少,经期紊乱”就诊于妇科门诊。当时查体:身高162cm,体重49kg,血压 130/85mmHg。乳房 Tanner Ⅱ期,无腋毛。外阴幼女型,Tanner Ⅰ期,在后期随访过程中,嘱其监测血压,于2021年发现血压逐渐增高,波动于130/70-145/95mmHg之间。2021年8月查体:身高162cm,体重51kg,血压145/95mmHg,乳房Tanner Ⅱ期,无腋毛。外阴幼女型,Tanner Ⅰ期。
1.2 方法
经过病史收集、体格检查和实验室辅检,初步考虑该患者为CAH。为进一步明确诊断,在取得患者及其父母知情同意的情况下,进行相关检测。
1.2.1 实验室检验:采集被检者空腹肘静脉血2ml,3 000转/min离心5min分离血清,采用化学发光法检测性激素、甲状腺功能(Atellica IM 全自动化学发光免疫分析仪)、肾素、醛固酮、ACTH和皮质醇(Autolumo A2000 化学发光检测仪);全自动生化分析仪检测肝肾功能和电解质(Siemens Advia 生化分析仪)。
1.2.2 影像学检查:采用美国GE LogicE9型超声仪对先证者及先证者姐姐行子宫及附件超声检查,采用美国GE680型64排螺旋CT机行肾上腺CT检查。
1.2.3 染色体核型分析:采用G显带技术对先证者及先证者姐姐行染色体核型分析,由本院检验科专业人员按相关规范手册要求进行检测分析。
1.2.4 DNA的提取和目的基因PCR扩增:抽取被检测者外周静脉血2ml,采用Qiagen FlexiGene DNA Kit 提取试剂盒(Qiagen公司)抽提DNA。北京康旭医学检验所使用带8bp index A01-H12 的引物(正向:CGC ACC TTG ATC TTC ACT TTG A;反向:GAA ACA GGG CTG TAT CTC TCC)扩增捕获的文库,对SureSelect-enriched DNA 文库进行 PCR 扩增。使用 AMPure XP(Beckman公司)磁珠纯化经扩增捕获的文库。使用 2100Bioanalyzer 仪器(美国 Agilent 公司)和 High Sensitivity DNA 试剂盒对 DNA质量和数量进行评估。
1.2.5 测序结果分析:纯化后的PCR产物使用NEXTSEQ 500 测序仪(美国Illumina 公司)进行检测,并用CASAVA(1.8.2)软件将原始数据转化为可识别的碱基序列,再经生物信息分析系统进行分析注释获得突变位点,并筛选出符合患者临床特征的位点,利用Sanger测序法进行一代验证和家系验证,明确变异来源。同时采用多重连接酶扩增反应技术(MLPA)检测患者CYP21A2,POR基因,以明确是否有大片段变异。
2 结 果
2.1 患者血液生化和激素水平
两名患者肝肾功能、甲状腺功能指标正常,先证者及先证者姐姐血钾水平均较正常参考值(正常值3.5-5.3mmol/L)略低。其余激素和醛固酮等检测结果见表1。
2.2 影像学检查
先证者:子宫附件超声(经直肠)示盆腔内未见子宫回声,未见明显卵泡回声;盆腔偏左右侧可见大小分别约为1.43cm×0.51cm,1.43cm×0.67cm低回声区(患者于2022年行手术切除该组织;病理检查为发育不良的睾丸及附睾组织);肾上腺CT结果显示:右侧肾上腺多发钙化灶(图1)。先证者姐姐:妇科超声显示,子宫偏小,子宫切面内径为3.78cm×2.54cm×3.42cm,双侧卵巢囊肿;肾上腺CT显示正常。

表1 患者相关激素实验室结果
2.3 染色体核型
先证者染色体核型为46XY,先证者姐姐染色体核型为46XX。见图2。
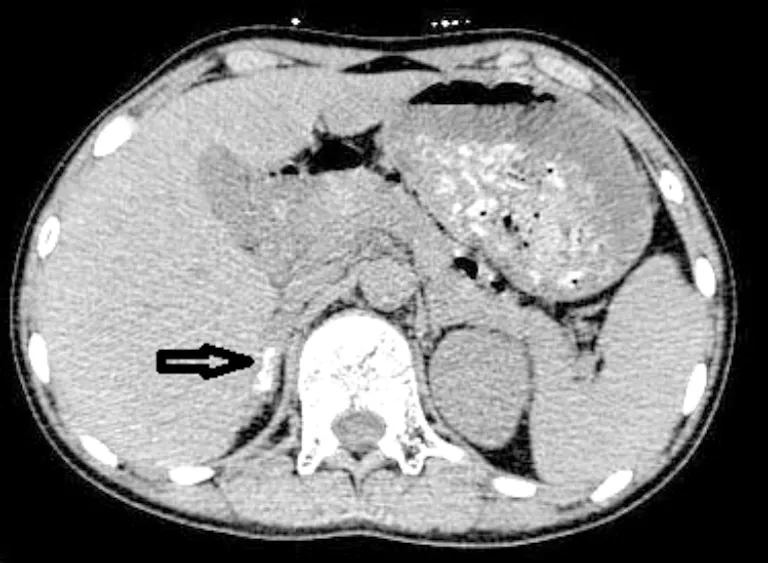
图1 先证者肾上腺CT影像(箭头处可见钙化灶)
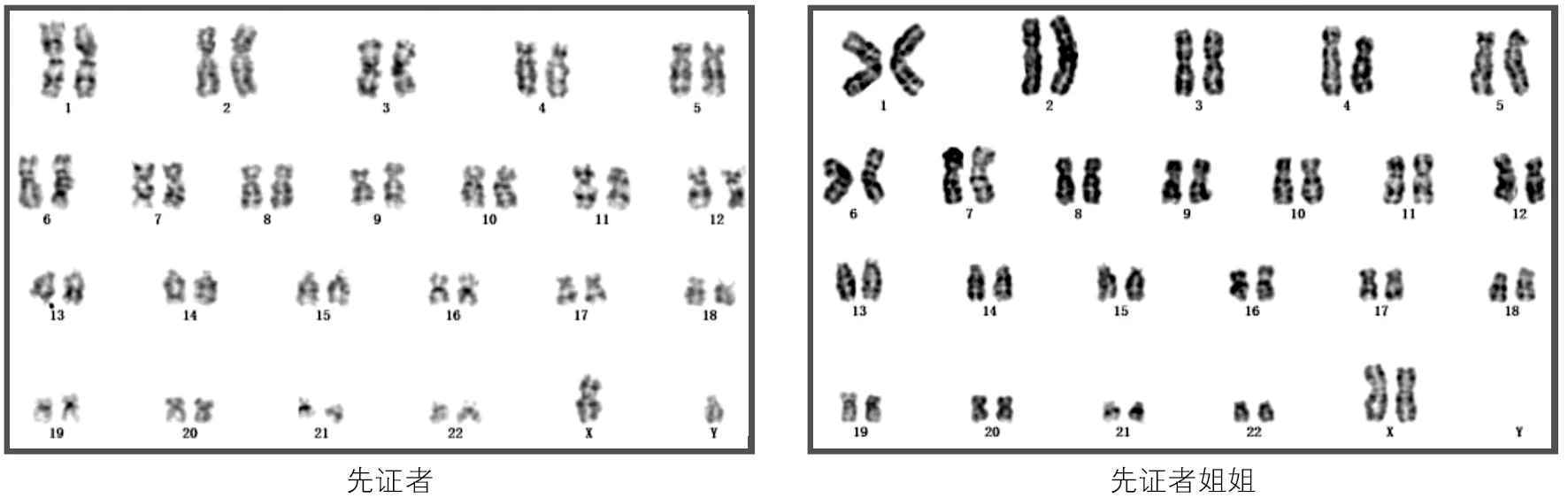
图2 两患者染色体核型分析
2.4 基因突变分析
经过基因测序对比分析,先证者及其姐姐CYP17A1基因第8外显子c.1283C>T(p. Pro428Leu)均发生纯合突变,该变异导致第428号氨基酸由脯氨酸(Pro)变为亮氨酸(Leu)。其父母均为c.1283C>T的杂合突变(图3)。经MLPA检测CAH相关基因,未发现受检者CYP21A2、POR基因存在大片段变异。17OHD患者家系图见图4。
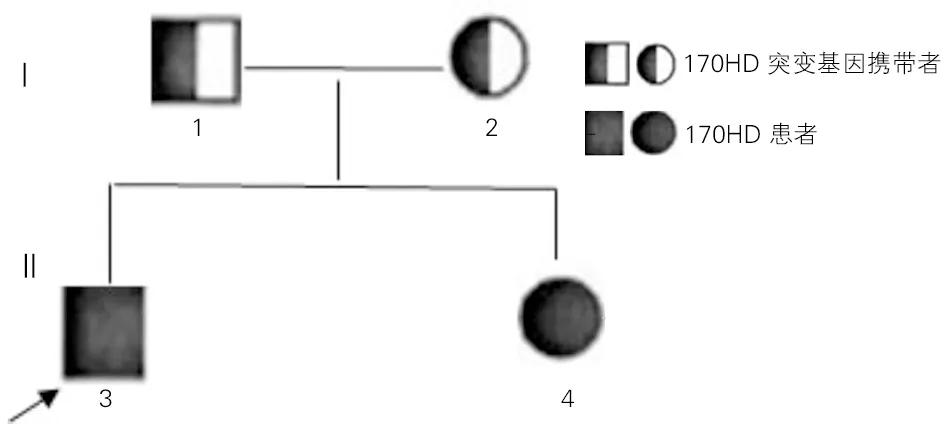
图4 17OHD患者家系图
3 讨 论
CYP17A1基因突变使细胞色素P450 17α-羟化酶(Cytochrome P450 17α-hydroxylase, P450c17)的活性发生不同程度缺失,随之阻碍类固醇的17α-羟化和17,20-裂解反应而致肾上腺皮质激素、性激素合成障碍[3]。 17OHD患者体内ACTH反馈性分泌增多,使双侧肾上腺皮质增生,并伴随某些中间代谢产物(如11-去氧皮质酮、孕酮等)过量蓄积,患者可出现低血钾、高血压、性发育异常等临床表现。Biglieri等[4]在1966年首次报告了该病。本研究中先证者正是以难以控制的高血压、低血钾就诊。理论上,11-去氧皮质酮具有极强的盐皮质激素作用,可导致水钠潴留,抑制RAS系统,降低醛固酮酶活性,从而使17OHD患者醛固酮水平下降[5]。但既往文献中对醛固酮水平的报道并不一致[6, 7]。本研究中两名患者醛固酮水平均在正常范围,其可能机制为17OHD患者的醛固酮并非如生理状态下由球状带产生,而是由束状带中衍生而来。此外,17OHD患者体内高水平的盐皮质激素前体物质可能干扰血液中醛固酮的检测[5]。
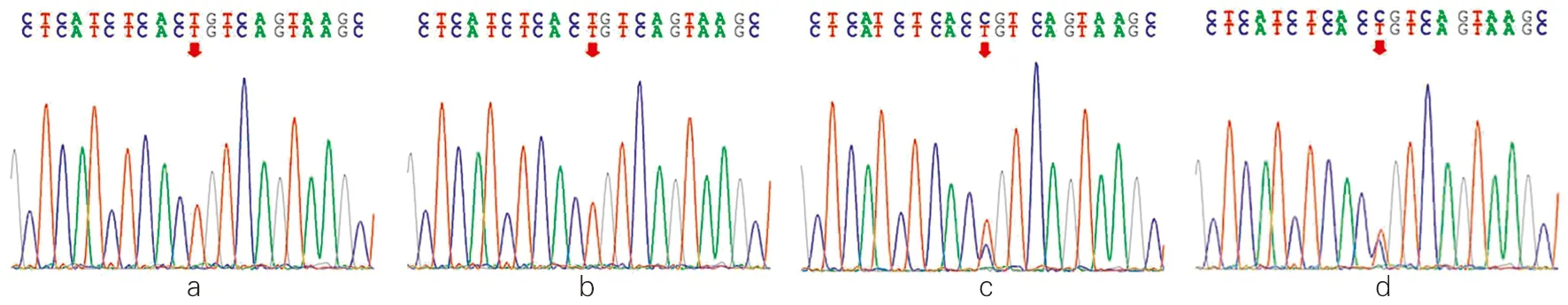
注:a为先证者(患者1),b为先证者姐姐(患者2),a和b均为CYP17A1基因c.1283C>T纯合突变,该变异导致第428号氨基酸由脯氨酸变为亮氨酸。c和d分别是患者父亲和母亲,其CYP17A1基因均为c.1283C>T杂合突变
由于P450c17(包含17α-羟化酶和17,20-裂解两种酶的功能)活性被破坏,雄烯二酮和硫酸脱氢表雄酮合成障碍,导致雌、雄激素水平降低,17OHD患者往往会有第二性征发育异常。本研究中先证者除了血压增高以外,还伴有性发育异常(无乳腺发育,阴道盲端,未见睾丸和阴茎。超声检查未见子宫)。由于核型46XY患者胚胎期发育正常的睾丸组织可分泌抗苗勒氏管激素,故患者没有子宫卵巢发育[8]。该类患者可能会因为睾丸未降而出现腹疝或腹股沟疝[9]。本研究中先证者幼年时曾行“腹股沟疝修补术”,后经手术证实是异位的睾丸组织。而46XX患者如果在青春期前没有明显的高血压或低血钾,她们往往以青春期延迟,原发性闭经或缺乏第二性征而就诊[9]。本文中的姐姐在疾病初期正是以“月经量少,经期紊乱”多次就诊于妇科。
CYP17A1基因位于10号染色体10q24.3,由8个外显子和7个内含子组成,编码508个氨基酸,主要在肾上腺和性腺中表达[3]。自1988年Kagimoto等[10]首次报道CYP17A1基因变异以来,至今已有100多种突变类型,包括错义突变、插入、缺失或剪切缺陷等[11]。约90%的中国病例基因突变发生于第6和第8外显子[12],其中,c.987C>A (p.Y329*)和c.1459_1467del (p.D487_F489)是中国患者常见的突变位点[13]。一项对24例巴西17OHD病例分析显示,c.1084C>T(p.R362C)和c.1216T>c(p.W406R)的突变率高达83%[14]。该现象可能与“祖先效应”有关。在本研究中,先证者和其姐姐的基因测序显示CYP17A1基因第8外显子均存在c.1283C>T纯合突变,导致428位脯氨酸突变为亮氨酸。其父母均为c.1283C>T杂合突变携带者(图4)。CYP17A1所编码的P450c17蛋白有4个重要的结构区域,分别是催化活性区、血红素结合区、底物结合区和氧化还原结合位点[15]。Costa-Santos[14]等采用CYP17A计算机模型分析蛋白质残基对酶活性的影响,发现P428位于的三维空间对血红素的结合和正确折叠均至关重要,突变的P428L可能直接影响血红素的结合,从而使17α-羟化酶/17,20-裂解酶活性丧失。
有趣的是,即使CYP17A1基因突变类型完全一致,但“姐妹”俩临床表现却有所差异。“先证者”以难以控制的高血压合并低血钾首诊,同时伴有性发育异常。而姐姐是以“月经不调”首诊,随着病程进展,2年后才逐渐表现出轻度高血压和血钾降低。性别对表型变异的影响可部分解释这种差异,相同CYP17A1基因突变的46XX患者的症状往往较46XY轻[16],其原因可能与雌二醇的高效力有关。有研究显示仅5%-8%的17,20-裂解酶活性即可促进第二性征的发育[14, 17]。但是,核型并非是影响临床表型的唯一因素。Kardelen等[18]发现,同样基因突变类型的患者,即使核型相同,但临床表现不尽一致。Espinosa-Herrera等[19]报道,两名厄瓜多尔姐妹均存在c.1216T>C(p.T406A)的纯合突变,其中姐姐有明显高血压和低血钾,但妹妹的血压和血钾均正常。由此可见,CYP17A1基因型和表型的相关性目前仍然未知,尚需进一步研究。
17OHD患者目前基本治疗方案是糖皮质激素补充治疗,旨在通过抑制ACTH过度分泌,减少中间代谢产物蓄积,以达到降低血压,纠正低血钾的目的。核型为46XY的患者需要尽早确定好将来的社会性别,提高患者的社会认同感,减轻其心理负担,并切除异位睾丸组织以免发生恶变。
综上所述,17OHD发病率低,患者临床表现复杂多变。临床医生应加强对本病的认识,避免误诊、漏诊。早诊断、早治疗可极大地改善患者生活质量。
◀
本文第一作者简介:
包 艳(1975-),女,汉族,博士,副主任医师,研究方向为内分泌与代谢性疾病
猜你喜欢
中华实用诊断与治疗杂志(2022年2期)2022-09-02
临床输血与检验(2022年3期)2022-06-22
热带作物学报(2020年9期)2020-10-29
中国现代医生(2020年12期)2020-07-04
郑州大学学报(医学版)(2019年3期)2019-06-03
心肺血管病杂志(2019年12期)2019-05-20
传染病信息(2019年2期)2019-05-17
中国继续医学教育(2016年18期)2016-02-15
江苏农业科学(2015年10期)2015-12-23
重庆医学(2015年12期)2015-03-05
