
PTPN11基因突变致泛发性雀斑样痣2例
2022-11-15 11:50黄闽嘉谢锦莹周顺婷
皮肤病与性病 2022年5期
黄闽嘉,吴 玮,谢锦莹,周顺婷,祝 玉
(广东医科大学附属医院皮肤性病科,广东 湛江 524000)
1 临床资料
患者,男,27岁,因“全身泛发性黑褐色斑点27年”就诊于我科门诊。患者出生时即发现全身弥漫分布黑褐色斑点,随年龄增长逐渐增多,颜色稍加深,与日晒无关,无自觉症状。既往体健,否认慢性病史及其余家族病史。父母非近亲结婚,家族中父亲为类似临床表现的患者(图1)。体格检查:各系统未见明显异常,头面部无畸形,未见眼距增宽、睑裂下斜和低位耳及耳后旋等,胸廓、脊柱无畸形,骨骼发育未见异常,外生殖器未见异常,未见精神发育迟缓。皮肤科检查:面颈部、躯干、四肢、掌跖区域等部位广泛分布大量圆形或椭圆形黑褐色斑点,直径(2~6 )mm,境界清楚,呈雀斑样改变,面部尤为密集,唇红部位有受累(图2),自颈部向下逐渐稀疏(图3 a~3f)。
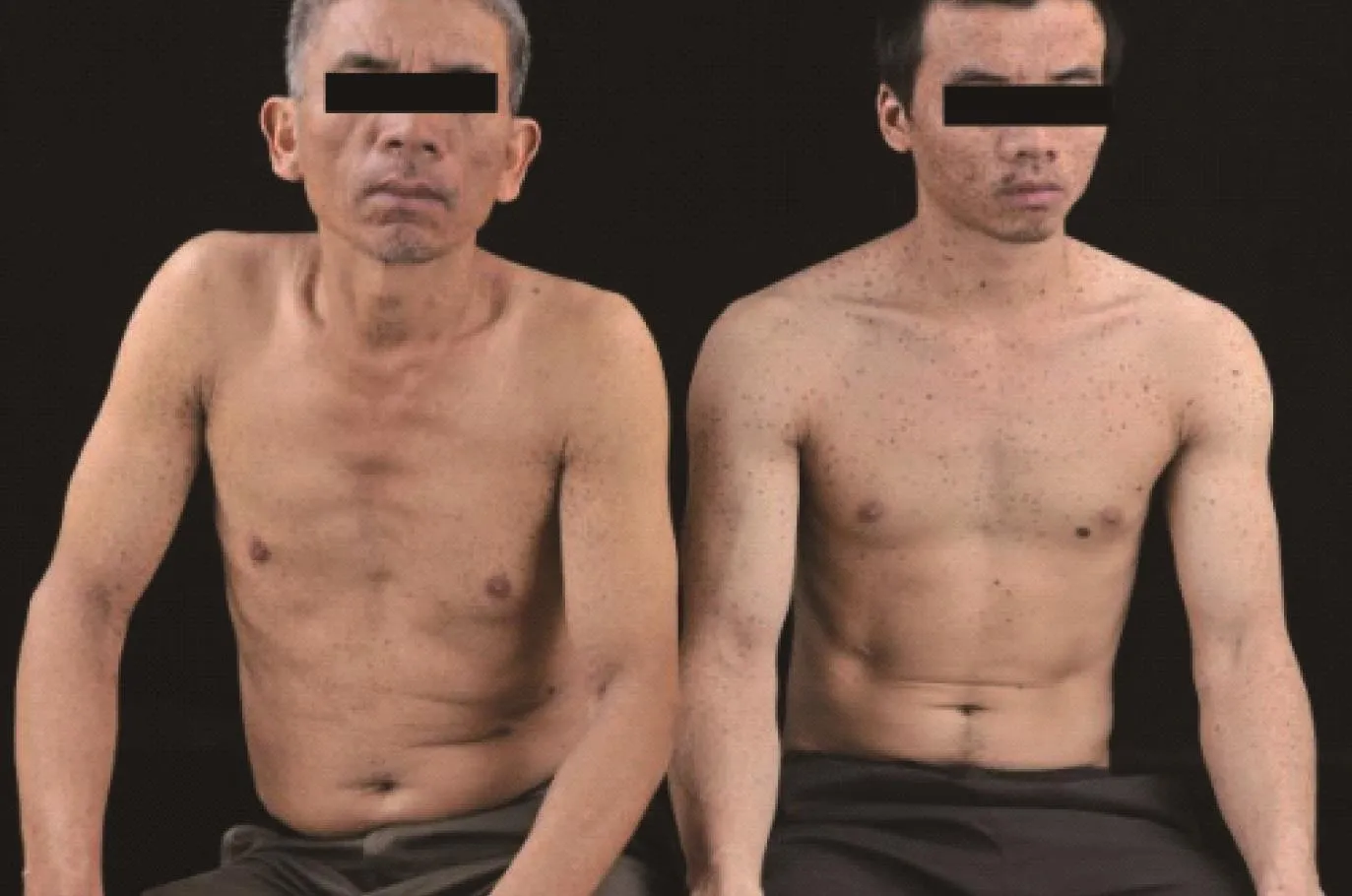
图1 患者及其父亲(左:父亲;右:患者)
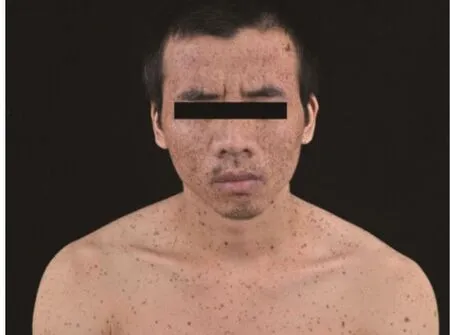
图2 面颈部、唇红部位受累
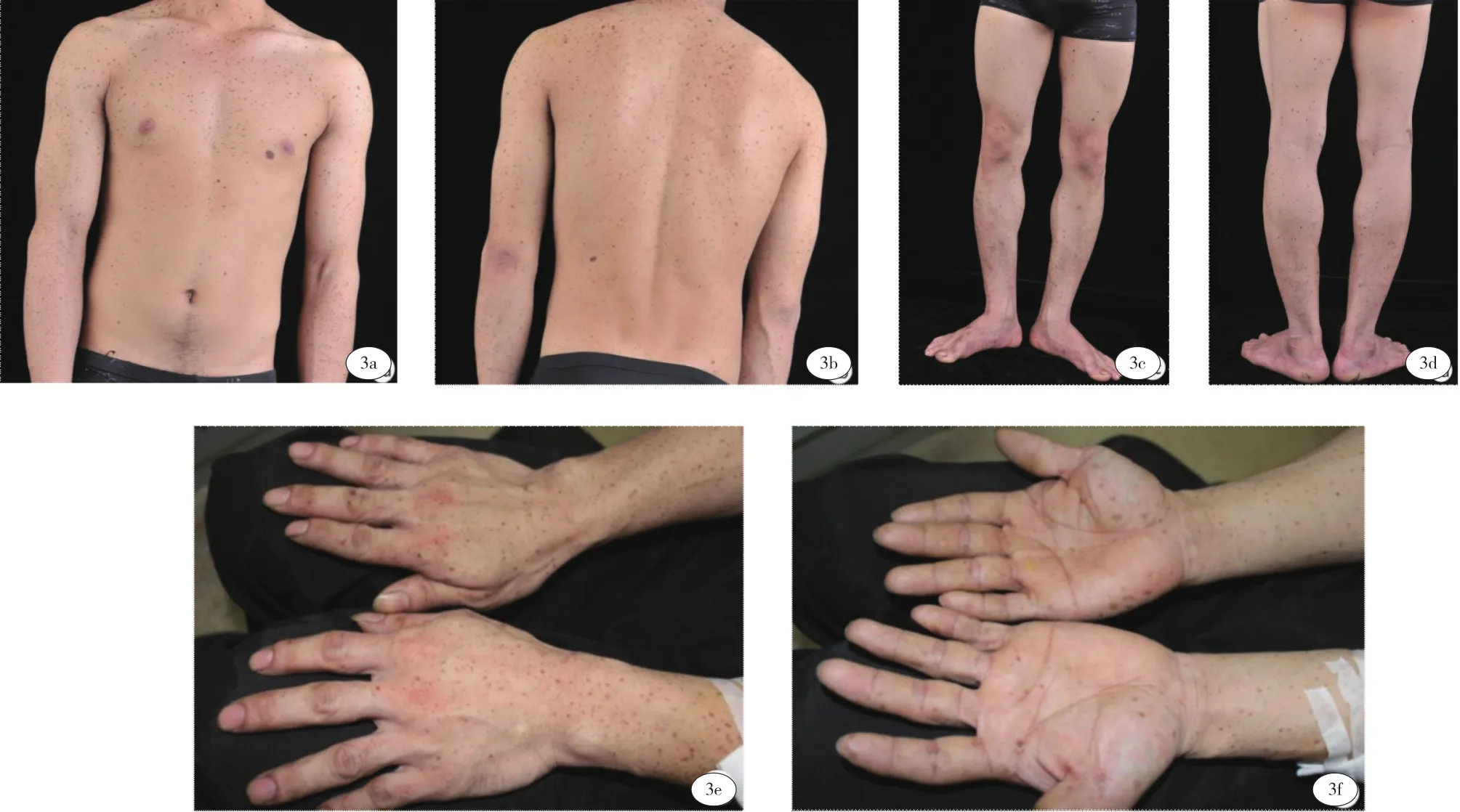
图3 (3a~3f)躯干、四肢、掌跖区域广泛分布的点状色素斑
实验室检查:经患者及其家属知情同意后,取先证者及其父亲外周血各5 ml,标本送至深圳华大临床检测中心,采用芯片捕获高通量方法进行DNA全外显子直接测序,使用Sanger测序法进行验证。先证者及其父亲的临床全外显子组检测-单人(B类)检出PTPN11;NM_002834.3:c.1493G >T(p.Arg498Leu)变异,在第13号外显子中发现了一个杂合错义突变,即G1493T转位导致氨基酸序列R498L改变(图4)。此突变位点曾在法国首次报道,可能参与了该家系患者发病的遗传学机制。

图4 患者(①)及其父亲(②)的PTPN11基因突变分析:PTPN11基因第13号外显子发生c.1493G>T(p.R498L)杂合错义突变。
诊断:泛发性雀斑样痣。
治疗:由于患者无自觉症状,体格检查也未见明显异常,故未行特殊处理。
2 讨论
泛发性雀斑样痣(generalized lentiginosis,GL,OMIM:150900)是以多发性黑子为突出表现的常染色体显性遗传病。目前临床上根据是否有皮肤以外其他系统受累分为发疹性黑子病(eruptive lentiginosis,OMIM:151001)和泛发性雀斑样痣综合征(multiple lentigines syndrome)即 LEOPARD 综 合征(LEOPARD syndrome,LS,OMIM:151100)两种类型。LS由Gorlin[1]等于1969年首先报道,其典型症状包括:多发性雀斑样痣、心电图异常、宽眼距、肺动脉狭窄、生殖器异常、生长迟缓和神经性耳聋。1976年Voron 等[2]提出该病诊断标准:① 有多发性雀斑样痣,且至少有两个其他基本特征。② 没有多发性雀斑样痣,但至少满足三个其他基本特征,同时患者有一个一级亲属确诊 LS。目前对于发疹性黑子仅在少数病例中检测到了SASH1 基因突变,而PTPN11、BRAF、RAFl 的基因突变涵盖了 95%[3]的LS 临床病例,其中约90%以上的患者由PTPN11突变所致[4]。目前已发现 LS相关的PTPN11错义突变15个,其中 Tyr279Cys和 Thr468Met约占 65%[3],且所有突变均位于PTPN11第7、12、13外显子内,其中外显子7和外显子12突变约占90%。
由于雀斑样痣的皮损通常在出生时或出生后不久即出现,一般都在成年以前发生,而内脏病变多数出现在成年后或者更晚,因此仅有皮疹的泛发性雀斑样痣作为一种单独的疾病存在还是综合征的一部分,目前尚有争议[5、6]。
本报道中的病例有如下特点:① 自出生起即有皮损,随年龄增大皮损增多,颜色加深,泛发全身并对称,掌跖皮疹略稀疏,口腔黏膜无皮疹;② 无系统损害;③ 具有遗传史,家族中父亲发病;④ 基因检测发现PTPN11基因第13号外显子发生c.1493G>T(p.R498L)杂合错义突变。该患者未行皮肤组织病理检查。综上所述,该病例从临床表现方面尚未达到LEOPARD综合征的诊断标准,但其致病基因及突变位点与LS密切相关,与魏露露等[7]收集的一个泛发性雀斑样痣家系相似,仅有皮肤表现,而无系统损害,因此考虑诊断为泛发性雀斑样痣。经文献检索发现该突变位点于2007年在法国首次报道[8],迄今为止国内尚无此基因突变的报道,因此丰富了我国汉族人LS 的遗传数据库。该突变及既往发现的所有LS致病突变均位于PTPN11的SHP-2结构域内,SHP-2在调控多种下游生物反应的媒介中起重要作用,LS突变体使 SHP-2 功能降低,导致多条细胞信号通路调控紊乱,影响细胞正常生长、发育、分化及凋亡而致病[9]。
泛发性雀斑样痣常伴随多个综合征出现,并作为综合征的一个重要组成部分,因此往往需要与多种疾病相鉴别。除外特征性的临床表现,基因检测和分析也有助于诊断和鉴别诊断。PTPN11基因突变亦见于 Noonan 综合征,但罕见雀斑样痣皮疹、咖啡斑、耳聋,可与 LS 鉴别。Carney 综合征为染色体 17q22-24 上的PRKAR1A 基因突变,是一种显性遗传性多发性内分泌肿瘤综合征,皮肤点状色素沉着(雀斑样痣、雀斑、咖啡斑、蓝痣),口腔黏膜及外阴黏膜部位的受累是本病的特征性表现[10]。色素沉着息肉综合征即Peutz-Jeghers综合征,为常染色体显性遗传,致病基因为LKBl/STKl1[11],临床特征包括皮肤黏膜雀斑样痣(眼、 鼻孔、唇缘、口腔、肠黏膜、手脚掌和肛周棕色或蓝色斑)、胃肠道错构瘤及早发肿瘤风险。Laugier-Hunziker综合征皮疹表现为手足指、掌跖部位、口唇和口腔黏膜部位多发的棕褐色斑,在50%的患者中可以见到表现为纵行黑甲的甲改变[12、13],本病不伴有系统受累。
泛发性雀斑样痣尚无特效治疗,因此对于暂无皮肤外病变者,随访及进行体格检查显得十分重要,需要评估心脏、泌尿生殖、骨骼、神经系统以及听力等,同时行基因分析。早期诊断和相关风险评估可改善预后。
猜你喜欢
今日农业(2022年15期)2022-09-20
广西医科大学学报(2022年5期)2022-06-07
昆明医科大学学报(2022年3期)2022-04-19
中老年保健(2021年12期)2021-08-24
皮肤病与性病(2021年3期)2021-07-30
当代水产(2020年4期)2020-06-16
中南医学科学杂志(2019年6期)2019-12-05
摄影之友(影像视觉)(2017年11期)2017-11-27
创新作文(小学版)(2017年24期)2017-02-25
实用皮肤病学杂志(2015年4期)2015-12-22
