
pAPN和NEU3的敲除对猪传染性胃肠炎病毒(TGEV)侵染的影响
2022-11-15 06:37:38李兆龙丰志华张冰晨梁旺旺陈文志
中国兽医学报 2022年10期
李兆龙,丰志华,张冰晨,方 舟,梁旺旺,陈文志
(1.福建省农业科学院 畜牧兽医研究所,福建 福州 350013;2.福建师范大学 南方生物医学研究中心,福建 福州 350013)
猪传染性胃肠炎病毒(transmissible gastroenteritis virus,TGEV)是猪传染性胃肠炎的病原,不同种类和不同日龄猪都呈现易感性。以往该病主要在冬季较为常见[1]。但近几年,随着养殖密度和规模增加,呈现一年四季皆有发病。它主要临床症状为呕吐、脱水和严重腹泻等。而中大猪和成年母猪感染后病死率低,但哺乳仔猪感染后发病率和病死率可达100%,是我国当前生猪养殖中的一种固疾,给畜牧业生产带来巨大的损失。为了进一步加强该病的防控,有必要深入研究TGEV的感染机制,为药物和疫苗开发,或抗病品种研发奠定基础[2]。之前研究报道,氨基肽酶(pAPN)是TGEV侵染的主要受体,猪体内的pAPN敲除后,明显抑制TGEV的侵染,减少TGEV的发病率[3-5]。但是,TGEV的侵染方式多样,pAPN敲除不足以完全抑制TGEV的感染[6]。此外,唾液酸神经氨酸酶(NEU3)作为病毒侵染的另一个重要中间体,发挥重要的作用[10-11]。病毒的侵染是一个复杂系统,详细的作用机理尚待进一步揭示。pAPN和NEU3共同敲除是否能更好地抑制该病毒的入侵,尚待进一步的研究。
本试验旨在运用CRISPR基因敲除技术,对ST细胞中的pAPN和NEU3双基因共同敲除,以评估它对TGEV的入侵影响,从而推动相关疫苗和抗TGEV品种猪的研究。
1 材料与方法
1.1 试验细胞、细菌和质粒ST细胞购上海英杰生物科技有限公司;pX459购自美国Addagen公司。
1.2 试剂本试验所用试剂及来源见表1。

表1 试剂及来源
1.3 引物及sgRNA为了敲除ST细胞中的pAPN和NEU3基因,参照sgRNA设计网站:https://zlab.bio/guide-design-resources进行双基因的sgRNA设计,并送测序公司合成。
1.4 试验设计见图1。本研究设计了针对pAPN 和NEU3两个关键基因CRISPR基因编辑系统,通过脂质体转染ST细胞,培养24 h后,用嘌呤霉素进行筛选,然后挑取单克隆细胞,测序验证。再将TGEV感染pAPN 和NEU3两个关键基因敲除的ST细胞,观察对细胞病变的改善、TGEV滴度的影响、干扰素的mRNA水平变化等指标,以评估它对TGEV侵染的影响。
1.5 重组编辑两目标基因载体的构建本研究中使用的sgRNA是在sgRNA Designer:CRISPRko (broadinstitute.org)网站上设计,针对pAPN 和NEU3两个关键基因设计多个sgRNA(表2),由福州铂尚生物科技公司合成,合成后的sgRNA退火,与酶切的Px459编辑质粒按相关体系组成进行连接,见表3。连接后转化至感受态细菌DH5a,然后挑取单克隆菌,进行PCR扩增并提DNA,将扩增出来的产物送福州铂尚生物科技公司测序验证。sgRNA退火体系:10×PCR 缓冲液2.5 μL,F-sgRNA 1 μL,R-sgRNA 1 μL,双蒸水20.5 μL, 共计25 μL。

表2 sgRNA名称及序列

表3 验证引物
将sgRNA的各组分依次加入后混匀,再低速离心20 s。将各反应物放置反应管进行反应,反应条件如下:初始温度 95℃,进行10 min;然后进行梯度温度处理:按95℃~85℃,2.5℃/s;85℃~25℃,0.25℃/s;25℃ 5 min。反应结束后取3 μL反应产物用1%琼脂糖凝胶电泳检测验证。双酶切体系:质粒(1 μg),10×Fast 缓冲液2 μL,快消化酶-F 1 μL,快消化酶-F-R 1 μL,双蒸水补足25 μL,总计25 μL。
将质粒所需各组分,按上表安排,依次加入后低速离心混匀20 s,后37℃维持15 min,取3 μL用1%琼脂糖凝胶电泳检测验证。连接反应体系:10×T4DNA缓冲液2.5 μL, sgRNA2质粒70 ng, T4DNA 连接酶1 μL,双蒸水补足25 μL,总计25 μL。
按连接试剂盒的说明进行常规的连接,再将连接产物转化到50 μL感受态细胞,于 37℃培养1 h,再3 000 r/min离心,再用20 μL重悬液,后均匀涂至含AMP的LB平板,置37℃倒置培养过夜。18 h后,挑取单克隆并加入2 mL LB,进行扩增培养14 h。然后吸取单克隆菌液300 μL 送至福州铂尚测序公司测序验证,将测序结果使用bioedit软件进行比对分析验证。
1.6 构建pAPN 和NEU3两个关键基因敲除ST细胞
1.6.1构建 将构建成功的重组编辑质粒Px459-pAPN和Px459-NEU3,按1∶2的比例加入脂质体LIPO2000,混匀并加入20 μL体积的无血清培养基。待6孔板ST细胞长至汇合度70%时,用无血清培养基清洗2次,然后加入转染混合液,37℃培养18 h。去除转染混合液,加入正常的血清培养液,并按100 mg/L加入嘌呤霉素进行筛选;4~7 d后,去除含嘌呤霉素的培养液,加入新鲜DMEM培养基,培养大概12 h后,放置显微镜观察,可见微小单克隆细胞群落,然后在镜下挑取单个克隆,放置新的培养皿,加入新的培养基进行扩大培养。扩大培养后收取克隆细胞,提取总DNA进行pAPN-/-和NEU3-/-双基因敲除PCR验证。然后利用Western blot在蛋白水平上验证pAPN-/-和NEU3-/-双基因敲除细胞株。
1.6.2PCR及Western blot验证 为了进一步验证单克隆细胞株是否成功敲除pAPN和NEU3双基因,我们设计了2对引物,进行PCR验证,引物序列如表3。细胞基因组DNA的提取见常规DNA提取试验盒操作(略)。PCR验证反应体系:5×PCR反应混合液(Mix)25 μL,模板5 μL,双蒸水20 μL。反应条件:42℃ 2 min,4℃保存。免疫印迹试验:配置10%的分离胶,并加入到制胶板底层,为了去除分离胶内的气泡并保持分离胶的平行,在加完分离胶前加入水压平放置15 min。用滤纸将分离胶上层的水吸干净,再缓缓加入5%的浓缩胶,并插入加样梳,放置40 min;接着轻缓拔除凝胶上的梳子,再重新把胶板放入电泳槽,添加一定比例的电泳缓冲液,当缓冲液没过长玻璃板后,停止添加。待液面静止5 min,用加样器往凝胶上样孔加入蛋白样品。然后打开电泳仪电源,设置恒压80 V 持续20 min,后调整电压至120 V持续约100 min;关闭电源,废弃上层的浓缩胶,再用刀片将含有目的蛋白和内参蛋白的凝胶切下放置4℃冰箱保存及待用。接着再根据本次凝胶大小来裁剪滤纸和PVDF膜,将胶、PVDF膜和滤纸浸入转膜缓冲液10~20 min后,先铺好3层滤纸,用玻璃管除去气泡,将胶水涂在去除气泡的滤纸上,然后将PVDF膜覆盖在胶水上,轻轻去除气泡,将靠近胶水的一侧指向黑板,靠近薄膜的一侧与白板对齐。组装改进的薄膜装置,再安装一个改进的膜,300 mA恒定电流至50 min;电转后,取膜,冲洗靠近胶水的一侧,用TBST(5×TBS稀释0.05%吐温-20)冲洗3次。封闭反应:室温下(25℃),用10%脱脂奶粉或5%BSA封闭PVDF膜1 h。抗原抗体反应:蛋白一抗按抗体规格比例加入,4℃摇床过夜孵育。 第2天,取出PVDF膜,用TBST冲洗3次,每次10 min,然后加入相应的荧光标记二抗,室温孵育2 h,再用TBST冲洗PVDF膜,每次10 min;每片扫描膜用双色红外激光成像仪观察(Odysey),CorelDRAW12软件分析绘图。
1.7 TGEV感染pAPN 和NEU3双基因敲除ST细胞分别取1.1×106个/mL ST细胞和pAPN和NEU3双基因敲除的ST细胞,分别放置4个6孔板,过夜培养。待细胞贴壁完成后,加入1.1×105TCID50/mL的TGEV,并分别于3,6,12,24,36,48 h在显微镜下观察并拍照记录细胞的病变情况。同时收集一孔细胞,提取细胞中的病毒RNA和细胞中干扰素的mRNA,然后实时定量验证TGEV的拷贝数变化、干扰素的mRNA水平及TGEV的纤连蛋白mRNA水平。RNA提取过程如下:按10 cm2/mL 比例加入Trizol,对培养贴壁的两种ST细胞进行消化、裂解,转移至离心管于室温放置5 min,使其充分裂解;Trizol和氯仿按1∶0.2比例添加,温和地混匀,再室温(25℃)放置15 min;然后在4℃离心机中按转速 12 000 r/min离心15 min;取出离心管,小心地吸取上层液体,将其转移至另一根干净的离心管中;再按1∶0.5的比例(Trizol∶异丙醇)加入异丙醇,上下轻轻摇混匀,室温25℃放置5~10 min; 再经4℃ 12 000 r/min转速离心10 min,小心弃上清,以1∶1 加入75%乙醇,温和地温匀;4℃ 8 000 r/min离心5 min,弃上清,室温干燥5~10 min.
1.8 Real-time PCR相对定量法检测基因的表达情况要求各引物的扩增效率在90%~110%之间,各个引物的扩增效率相差不超过5%。用不同引物对模板进行PCR,将PCR产物纯化后导入T载体。然后以108拷贝为起始浓度,依次10倍稀释5个梯度,摸索出最佳PCR扩增条件,再对样本进行相对定量。Real-time PCR反应系列:SYBR®Premix Ex Taq Ⅱ(Tli RNaseH Plus) ROX Plus 10 μL,上游引物(10 μmol/L)0.4 μL,下游引物Reverse Primer(10 μmol/L)0.4 μL,mRNA溶液(30 mg/L) 3 μL,双蒸水ddH2O 7.2 μL,总计20 μL。反应条件: 95℃ 30 s;95℃ 5 s,55℃ 30 s,72℃ 30 s,40 cycles。熔解曲线阶段:95℃ 15 s,72~88℃,每个周期0.1℃递增。所用实时荧光PCR引物见表4。

表4 实时定量PCR引物
2 结果
2.1 pAPN 和NEU3双基因敲除ST细胞挑取经嘌呤霉素筛选的pAPN 和NEU3双基因敲除ST单克隆细胞,提取细胞的基因组,进行PCR验证,结果产生预期的2个条带,分别为2 881,1 349 bp,与预期的引物结果一致(图2)。然后将该PCR产物测序,结果证实分别在pAPN 和NEU3的245和227 bp 位置出现碱基缺失。用pAPN 和NEU3的双抗,进行Western blot验证,野生型细胞完全能孵出预期的条带,但敲除株的ST细胞在预定位置并没有明显的条带。结果证实pAPN 和NEU3双基因在ST细胞中成功敲除,该基因的蛋白表达或受到抑制,或结构遭破坏。
由图2可知,在单克隆细胞中,吸取200 μL提取基因组DNA,然后进行PCR反应,结果在2 881和1 349 bp位置出现了条带,证明筛选得到了双基因敲除的细胞株。

M.DL5000 DNA Marker;1,2.阴性克隆;3,4.阳性克隆
由图3可知,将 2 881,1 349 bp位置出现的条带测序,结果证实245,227 bp位置出现碱基缺失。

图3 pAPN 和 NEU3双基因敲除测序结果
由图4可知, 取245,227 bp位置出现碱基缺失的ST细胞,进行蛋白免疫印迹试验,敲除株的ST细胞pAPN蛋白表达明显减弱,而NEU3完全看不到。

图4 pAPN和NEU3的敲除及免疫印迹印证
2.2 TGEV感染pAPN 和NEU3双基因敲除ST细胞对细胞的影响将1.1×105TCID50/mL的TGEV感染pAPN和NEU3双基因敲除的ST细胞和野生型ST细胞,6 h后,野生型ST细胞出现部分细胞病变,感染TGEV 12 h,野生型细胞出现明显的细胞聚集和死亡的细胞病变,24,36 h后,野生型细胞的聚集、拉网和死亡的细胞病变现象比双基因敲除的ST细胞更为明显(图5)

图5 TGEV感染pAPN 和NEU3双基因敲除ST细胞和野生型ST细胞
2.3 TGEV感染pAPN 和NEU3双基因敲除ST细胞明显下调了干扰素和纤连蛋白的产生Beta干扰素是机体产生的一类糖蛋白,它具有高度的种属特异性。当机体感染病毒时,会产生大量的干扰素来抵制病毒的增殖。因此,在本试验中,通过干扰素的mRNA水平,间接地反映初期病毒的强弱,当前期感染病毒较强,激发机体的免疫反应就大,产生的Beta干扰素水平就较高。pAPN和NEU3双基因敲除ST细胞,产生的干扰素水平远低于野生型细胞,这个结果间接说明敲除细胞内的病毒量可能低于野生型细胞(图6)。
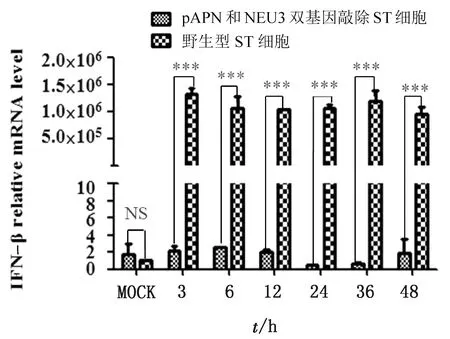
图6 干扰素mRNA
实时定量PCR检测,TGEV感染pAPN和NEU3双基因敲除ST细胞,产生的Beta干扰素mRNA远低于野生型细胞,差异极显著(*P<0.05, ***P<0.001)。
TGEV属于冠状病毒科冠状病毒属,病毒直径为90~200 nm,它的主要结构蛋白是纤突(S)蛋白、包膜(E)蛋白、膜(M)蛋白和核衣壳(N)蛋白。因此,我们将细胞中TGEV的纤突(S)蛋白的mRNA作为TGEV侵染细胞的标志。结果表明,野生型细胞感染TGEV后,细胞中病毒的S蛋白的mRNA在108,而pAPN和NEU3双基因敲除ST细胞S蛋白的mRNA还不足102,远低于野生型细胞,差异极显著。结果表明pAPN和NEU3双基因敲除ST细胞可能抑制了病毒的入侵(图7)。
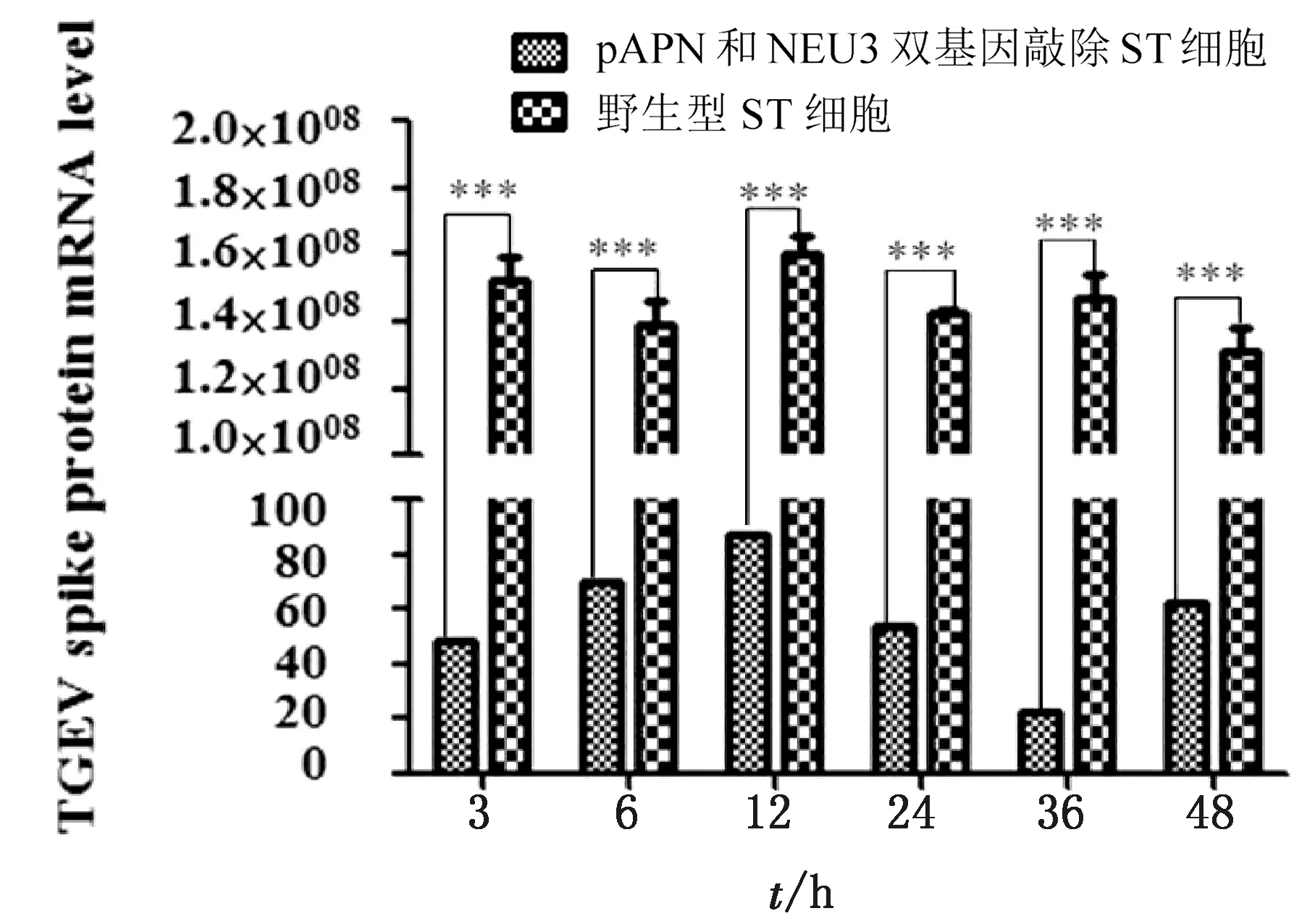
图7 TGEV的S蛋白的mRNA
实时定量PCR检测,TGEV感染pAPN和NEU3双基因敲除ST细胞,产生的S蛋白mRNA远低于野生型细胞,差异极显著(*P<0.05, ***P<0.001 )。
2.4 不同滴度的TGEV感染pAPN 和NEU3双基因敲除ST细胞为进一步验证pAPN 和NEU3双基因敲除ST细胞可以抑制TGEV的入侵,用不同滴度的TGEV侵染pAPN 和NEU3双基因敲除ST细胞,结果发现和对照组细胞相比,在2.2×104,4.4×104,4.4×105,8.8×105TCID50/mL的细胞病变明显较轻,并且随着滴度增加,敲除细胞并没有明显的变化。而对照组细胞,随着滴度上升,细胞病变更为明显(图8)。

图8 不同滴度的TGEV感染pANA和NEU3敲除ST细胞和野生型细胞
3 讨论
有研究证实,pAPN是TGEV侵染机体的主要受体,将猪体内的pAPN敲除,会明显抑制TGEV的侵染,减少TGEV的发病率[7-10]。但是,TGEV的侵染方式多样,pAPN敲除不足以完全抑制TGEV的感染。NEU3作为病毒侵染的另一个重要中间体,发挥了重要的作用[10-11]。本研究结果证实,将pAPN和 NEU3双敲的ST细胞,不管是病毒主要结构蛋白,还是病毒感染引起的Beta干扰素的水平,都明显比对照野生型低,细胞的病变也不明显。此外,我们用不同滴度的TGEV侵染双基因敲除细胞,结果发现,它的细胞病变并不随着感染滴度的增加而变化。此结果从另一个层面说明,pAPN和NEU3双敲,对TGEV的侵染有更为明显的抑制,但对病毒滴度的变化,还有待进一步研究。
本研究中,将纤突蛋白作为病毒在体内滴度变化的主要指征,主要基于TGEV主要结构蛋白是纤突(S)蛋白、包膜(E)蛋白、膜(M)蛋白和核衣壳(N)蛋白[12]。其中,以S蛋白尤为重要,也常作为病毒变异或毒力变化的指征[13]。
这个在人的冠状病毒中,尤为多见。近来发生的新冠病毒,大多以S蛋白作为研究的核心,不管是从检测还是疫苗的研究等。因此,在本研究将S蛋白的mRNA作为TGEV侵染细胞的标志。从细胞病变以及Beta干扰素的结果也证实,S蛋白作为病毒在体内滴度变化的主要指征,是能正确反映病毒侵染细胞的情况。
此外,我们将Beta干扰素作为病毒侵染细胞初期情况的其中一个指征,也是基于大部分病毒侵染机体或细胞,都会激起机体强烈的免疫反应,产生大量的Beta干扰素[14]。病毒滴度越高或毒力越强,产生的Beta干扰素量就越大。因此,它在一定程度,反映了病毒侵染细胞的强弱和病毒滴度的高低。但它并不能替代病毒的滴度测定,它只能间接反映病毒的入侵情况。
本研究结果显示,pAPN和 NEU3双敲的ST细胞可明显减少TGEV入侵,病毒的拷贝数明显低于对照组,并且病毒滴度的增加对细胞的病变改变不明显;因此pAPN和 NEU3双敲,可为将来抗TGEV猪的选育和抗TGEV的药物筛选提供理论基础。
猜你喜欢
当代水产(2022年1期)2022-04-26 14:35:30
昆明医科大学学报(2022年2期)2022-03-29 00:51:20
华侨大学学报(自然科学版)(2021年4期)2021-07-30 02:18:58
中国保健营养(2021年16期)2021-04-03 18:15:56
中国现代医药杂志(2020年10期)2020-12-14 07:20:14
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
西南农业学报(2016年5期)2016-05-17 05:42:33
西南农业学报(2016年6期)2016-04-16 05:12:51
医学研究杂志(2015年3期)2015-06-10 06:41:52
特产研究(2015年1期)2015-04-12 06:36:20
