
动物肠道内容物中8种酸性低聚糖的液相色谱-质谱联用测定方法
2022-11-15 07:54许丽卫孙德慧耿梅梅贺志雄王久荣
动物营养学报 2022年10期
许丽卫 孙德慧 耿梅梅 陈 闻 贺志雄 王久荣
(中国科学院亚热带农业生态研究所,亚热带农业生态过程重点实验室,长沙 410125)
功能性低聚糖是最主要的一类益生元,研究表明,饲粮中添加功能性低聚糖可以提高动物肠道的免疫能力,维护机体健康[1-2]。功能性低聚糖在动物胃肠道既不能被水解也不能被吸收,可黏附肠道中的有害病菌,刺激肠道有益菌的繁殖代谢,调节肠道微生物菌群平衡,从而提高宿主的机体健康水平,降低由于消化道疾病导致的幼龄动物的死亡率[3-4]。酸性低聚糖是反刍动物初乳的重要组成部分,占比80%~90%,与生物体的炎症、癌症发生、新陈代谢以及神经系统的发育和维持等息息相关[5-6]。研究酸性低聚糖在幼龄反刍动物肠道内容物中的变化情况,对于揭示其对幼龄反刍动物肠道健康的作用机制具有重要意义。
酸性低聚糖的检测难点在于它们具有极强的亲水性与相似的化学结构,异构体较多,且没有发色官能团或荧光官能团。目前,酸性低聚糖含量的测定方法主要有:核磁共振法、凝胶排阻色谱法、高效阴离子色谱分析法、毛细管电泳法、反相和亲水排斥作用液相色谱法[7]以及液相色谱-质谱联用法[8]。核磁共振法所需样品量大,分析较为复杂;凝胶排阻色谱法较为耗时,不能将异构体完全分开,且需与其他手段联用后方可对目标物进行定量分析;高效阴离子色谱分析法虽有较高的灵敏度,但需要与凝胶排阻色谱法联用,按性质分类后分离测定;反相和亲水排斥液相色谱法需将目标物进行衍生后方可测定。目前,液相色谱-质谱联用法已逐渐成为分离酸性低聚糖的主要手段,其中石墨化碳液相色谱、荧光标记液相色谱或者不衍生直接进样的液相色谱法等[8-10]与质谱联用已用于分离酸性低聚糖,测定样品种类主要集中在人乳、牛乳、羊乳以及婴儿配方奶粉,色谱柱主要为氨基柱和亲水排斥色谱柱,离子模式为正离子或者负离子模式,测定的种类有3′-唾液基-3-岩藻基乳糖(3′-sialyl-3-fucosyl lactose,3′-S,3-FL)、3′-唾液乳糖(3′-salivary lactose,3′-SL)、6′-唾液乳糖(6′-salivary lactose,6′-SL)、唾液基乳糖-N-四糖a(sialyl lactose-N-tetrasaccharide a,LSTa)、唾液基乳糖-N-四糖b(sialyl lactose-N-tetrasaccharide b,LSTb)、唾液基乳糖-N-四糖c(sialyl lactose-N-tetrasaccharide c,LSTc)、双唾液基乳糖-N-四糖(disasialyl lactose-N-tetrasaccharide,DSLNT)、二唾液酰乳糖(disasialyl lactose,DSL)、6′-唾液基-N-乙酰化乳糖(6′-sialyl-N-acetylated lactose,6′-SLN)、3′-唾液基-N-乙酰乳糖胺(3′-sialyl-N-acetylated lactose,3′-SLN)。
目前,关于动物肠道内容物中酸性低聚糖测定方法研究较少。Amanda等[11]采用液相色谱-质谱联用法,在负离子、多反应监测模式下,对新生的荷斯坦公牛空肠、回肠以及结肠中的3′-SL、6′-SLN、3′-SLN、DSL和6′-SL共5种酸性低聚糖进行了测定,发现其在肠道内容物中的含量较低,为20~300 μg/g;但未见对LSTa、LSTc和3′-S,3-FL定量分析的报道。动物不同肠道内容物的含水量不同,且含有丰富的脂类、蛋白质等物质,导致肠道内容物样品基质复杂且均一性较差,准确定量酸性低聚糖较为困难。因此,本研究拟采用液相色谱-质谱联用法,建立一种简单、操作性强、准确、灵敏度高,且对动物不同肠段内容物中8种酸性低聚糖进行单次分离分析并绝对定量检测的技术。
1 材料与方法
1.1 试剂与材料
标准品:3′-S,3-FL(纯度>95%,法国Elicityl公司)、LSTa(纯度≥90%,法国Elicityl公司)、3′-SL(纯度≥95%,加拿大TRC公司)、6′-SL(纯度≥95%,加拿大TRC公司)、LSTc(纯度>90%,Glycosci)、DSL(纯度≥90%,Glycosci)、6′-SLN(纯度≥95%,英国Dextra公司)、3′-SLN(纯度≥95%,英国Dextra公司)。乙腈、甲醇(色谱纯,美国Fisher公司),乙酸铵(色谱纯,天津科密欧化学试剂有限公司),水(中国科学院亚热带农业生态过程重点实验室自制、电阻率为18.2 MΩ);氨基固相萃取小柱(100 mg,1 mL,深圳逗点生物技术有限公司);0.22 μm尼龙滤膜(天津市希波氏科技有限公司);样品为中国科学院亚热带农业生态研究所畜禽健康养殖研究中心提供的羔羊肠道内容物,-80 ℃保存备用。
1.2 仪器与设备
Exion LC 30A-Q-Trap 5500超高效液相色谱-串联三重四极杆-线性离子阱质谱仪(liquid chromatography-mass spectrometry,LC-MS/MS)(美国AB SCIEX公司),配备电喷雾离子源、自动进样器;色谱柱Acquity UPLC®BEH Amide(1.7 μm,100 mm×2.1 mm)及其配套保护柱(美国Waters公司);Vortex-6涡旋混合器(海门市其林贝尔仪器制造有限公司);ME104、XPR2电子天平(瑞士梅特勒-托利多公司);Classic DI超纯水系统(英国Elga公司);CR22 GⅡ高速离心机(日本日立公司)。
1.3 色谱和质谱条件
色谱部分。流动相:A相,15 mmol/L乙酸铵水溶液;B相:15 mmol/L乙酸铵乙腈:水(体积比9∶1),流速为0.3 mL/min;多次优化后,得到梯度洗脱条件为0~1 min,5% A相;1~2 min,25% A相;2~3 min,35% A相;3~6 min,35% A相;6~7 min,50% A相;7~8 min,50% A相;8.01~11 min,5% A相;总运行时间为11 min,进样体积3 μL,柱温40 ℃。
质谱部分。负离子模式,离子源电压为-4 500 V,离子源温度为500 ℃,气帘气为高纯氮气,压力为35 psi;碰撞气级别为中,Gas1和Gas2压力均为55 psi,多反应监测模式检测。8种目标物的多反应监测参数见表1。
1.4 样品前处理条件的优化
将采集的羔羊空肠内容物样品冷冻干燥,制成粉状样,分别对提取溶剂体积、溶剂提取次数、沉淀蛋白溶剂、沉淀蛋白溶剂体积、沉淀蛋白时间以及氨基固相萃取柱净化条件进行优化。
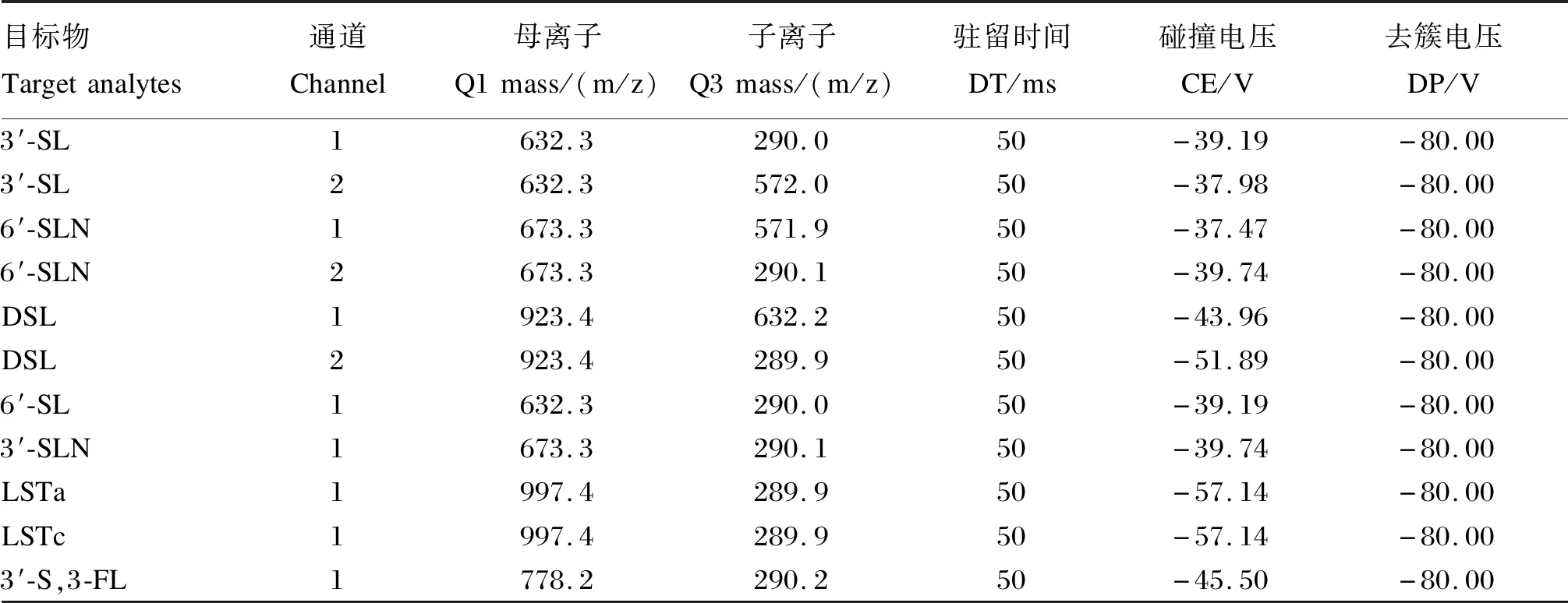
表1 8种目标物的多反应监测参数Table 1 Multiple reaction monitoring parameters of eight target analytes
通道1为定量离子对,通道2为定性离子对。The channel 1 was the quantitative ion pair, and the channel 2 was the qualitative ion pair.
1.4.1 提取溶剂体积的筛选
目标物水溶性好,因此选用水作为提取溶剂。精确称取每份样品0.050 0 g,加入水的体积分别为500、750、1 000、1 250和1 500 μL,涡旋混合均匀,超声提取10 min,每个处理3个重复;接着以16 400×g、4 ℃离心10 min;取上清200 μL,加入800 μL乙腈沉淀蛋白;再次以16 400×g、4 ℃离心10 min;取上清液过0.22 μm滤膜,按照1.3中的色谱和质谱条件上机测试。
1.4.2 溶剂提取次数的筛选
精确称取每份样品0.025 0 g,分别加入625 μL水,涡旋混合均匀,超声提取10 min,每个处理3个重复;分别提取1、2和3次,3次提取液均按照1.4.1中离心并沉淀蛋白,取上清液过0.22 μm滤膜,按照1.3中的色谱和质谱条件上机测试。
1.4.3 沉淀蛋白溶剂的筛选
精确称取样品0.050 0 g,加入1 250 μL水,涡旋混合均匀,超声提取10 min,分别提取2次;按照1.4.1中离心后,合并2次上清液。各取上清液200 μL,分别加入600 μL乙腈、600 μL甲醇2种常用的沉淀蛋白溶剂,每个处理3个重复;沉淀0.5 h,再次以16 400×g、4 ℃离心10 min;取上清液过0.22 μm滤膜,按照1.3中的色谱和质谱条件上机测试。
1.4.4 沉淀蛋白溶剂体积的筛选
精确称取样品2份,每份0.050 0 g,分别加入1 250 μL的水,涡旋混合均匀,超声提取10 min,以16 400×g、4 ℃离心10 min;提取2次,合并所有上清液;各取上清液200 μL,分别按照样品与沉淀试剂体积比为1∶1、1∶2、1∶3、1∶4和1∶5加入乙腈沉淀蛋白,每个处理3个重复;沉淀0.5 h,再次以16 400×g、4 ℃离心10 min;取上清液过0.22 μm滤膜,按照1.3中的色谱和质谱条件上机测试。
1.4.5 沉淀蛋白时间的筛选
精确称取样品3份,每份0.050 0 g,分别加入1 250 μL水,涡旋混合均匀,超声提取10 min,以16 400×g、4 ℃离心10 min;分别提取2次,合并所有上清液。各取上清液200 μL,加入乙腈600 μL沉淀蛋白,分别沉淀0.5、1.0、2.0、4.0、6.0和24.0 h,再次以16 400×g、4 ℃离心10 min,每个处理3个重复;取上清液过0.22 μm滤膜,按照1.3中的色谱和质谱条件上机测试。
1.4.6 氨基固相萃取小柱净化条件
1.4.6.1 淋洗液的筛选
固相萃取柱选择氨基柱(100 mg,1 mL),先用1 mL甲醇活化,然后取沉淀蛋白后的上清液500 μL上样,弃去流出液,分别利用乙腈∶水(体积比70∶30)和乙腈∶水(体积比50∶50)500 μL进行淋洗,淋洗液过0.22 μm滤膜,按照1.3中的色谱和质谱条件上机测定。
1.4.6.2 洗脱溶剂的筛选
固相萃取柱选用氨基柱(100 mg,1 mL),先用1 mL甲醇活化,取沉淀蛋白后的上清液500 μL上样,弃流出液,再分别用洗脱液1(500 μL水),洗脱液2(250 μL水、250 μL 15 mmol/L的乙酸铵水溶液)以及洗脱液3(500 μL 15 mmol/L的乙酸铵水溶液)洗脱,收集洗脱液,分别加入500 μL乙腈混合均匀后,过0.22 μm滤膜,按照1.3中的色谱和质谱条件上机测定。
1.5 标准工作曲线配制、线性范围及检出限
称取3′-SL、6′-SLN、LSTa、3′-S,3-FL、LSTc、3′-SLN、DSL、6′-SL的标准品,用乙腈:水(体积比50∶50)溶解,浓度分别为1 048、1 000、940、100、500、1 000、500和1 070 μg/mL的单种标准储备液。取各个标准物质的储备液,配制3′-SL、6′-SLN、LSTa、3′-S,3-FL、LSTc、3′-SLN、DSL和6′-SL浓度分别为50.00、2.50、2.35、2.50、5.00、2.00、3.00和16.10 μg/mL的混合标准溶液A,以此作为母液,配制一系列标准曲线,各标准曲线的样品浓度见表2。目标化合物根据峰响应强度进一步稀释测定其定量限和检出限。以各目标物的浓度为横坐标,峰面积为纵坐标作图,得线性回归方程;以目标物3倍信噪比的浓度为其检出限,10倍信噪比的浓度为其定量限。
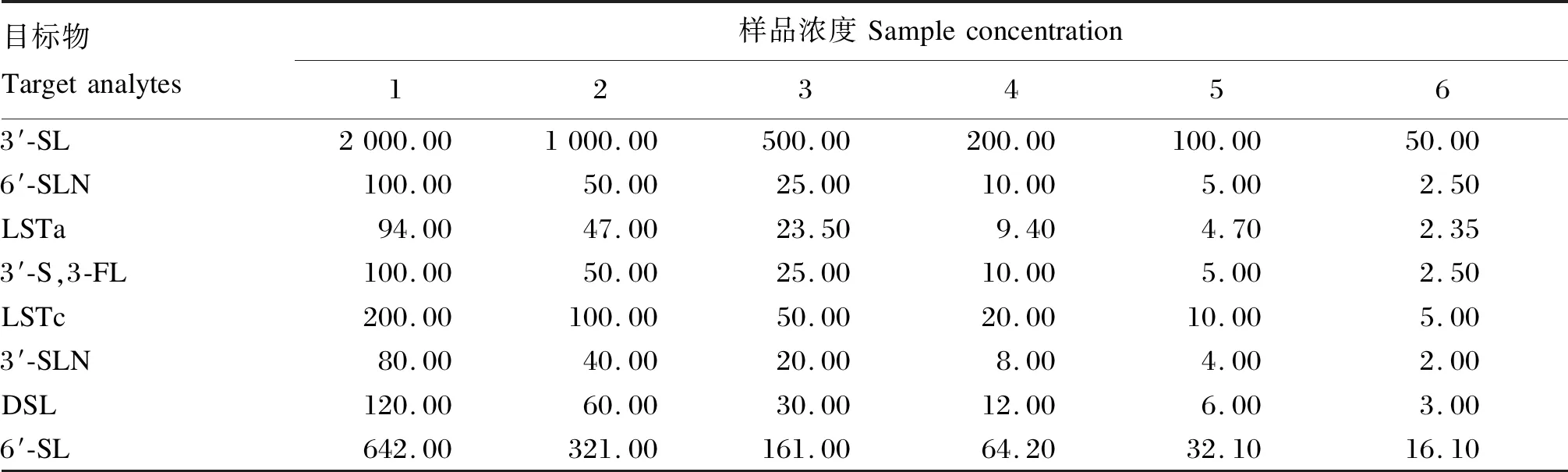
表2 标准曲线的样品浓度Table 2 Sample concentration of standard curve ng/mL
1.6 方法学考察
以样品浓度5的标准样作为供试品,重复测定7次,考察所建立方法的精密性;以羔羊十二指肠内容物为供试品,平行取样3份,按照优化前处理条件处理,考察所建立方法的重复性;以羔羊十二指肠内容物为供试品,分别加入20、40和60 μL的混合标准溶液A,按照优化的前处理条件处理,测定样品的加标回收率。
1.7 数据统计分析
试验数据采用Graphpad Prism 9.0软件进行统计分析,结果用平均值±标准差表示,图由软件直接生成。
2 结果与分析
2.1 提取溶液体积的优化
由图1可知,空肠内容物中未检出3′-S,3-FL、LSTa和LSTc;综合比较已检出的目标物发现,1 250 μL水提取的目标物峰面积最高。因此,在0.050 0 g样品中加入1 250 μL水进行提取,即样品(g)∶水(mL)=1∶25。
2.2 溶剂提取次数的筛选
由图2可知,第2次提取比第1次提取得到的目标物明显减少,第3次提取溶剂中几乎检测不到目标物。因此,提取次数确定为2次。
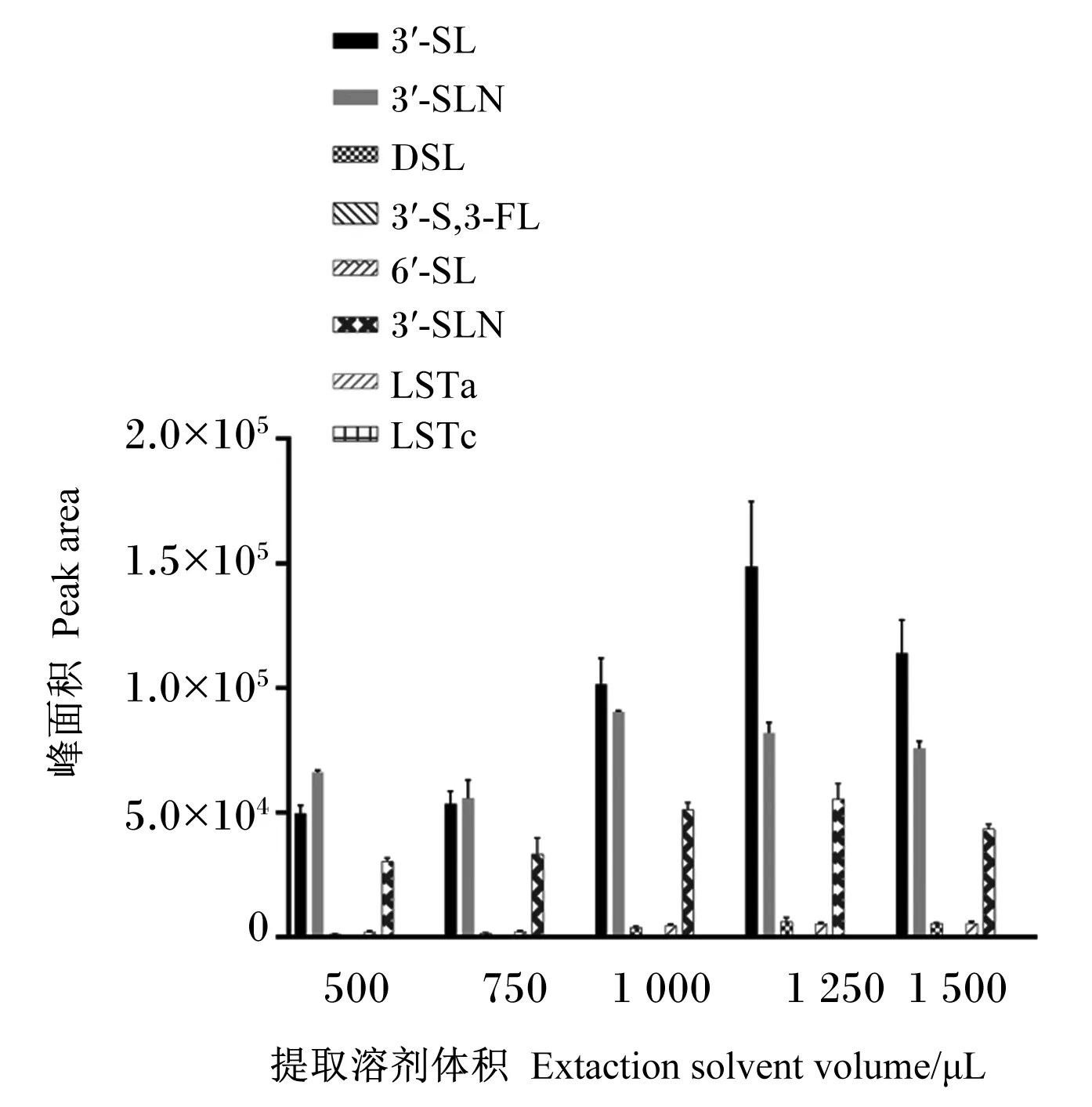
3′-SL:3′-唾液乳糖 3′-salivary lactose;6′-SLN:6′-唾液基-N-乙酰化乳糖6′-sialyl-N-acetylated lactose;DSL:二唾液酰乳糖 disasialyl lactose;6′-SL:6′-唾液乳糖 6′-salivary lactose;3′-SLN:3′-唾液基-N-乙酰乳糖胺 3′-sialyl-N-acetylated lactose;LSTa:唾液基乳糖-N-四糖a sialyl lactose-N-tetrasaccharide a,LSTa;LSTc:唾液基乳糖-N-四糖c sialyl lactose-N-tetrasaccharide c;3′-S,3-FL:3′-唾液基-3-岩藻基乳糖 3′-sialyl-3-fucosyl lactose。下图同 the same as below。图1 提取溶剂体积对目标物峰面积的影响Fig.1 Effects of extraction solvent volume on peak area of target analytes
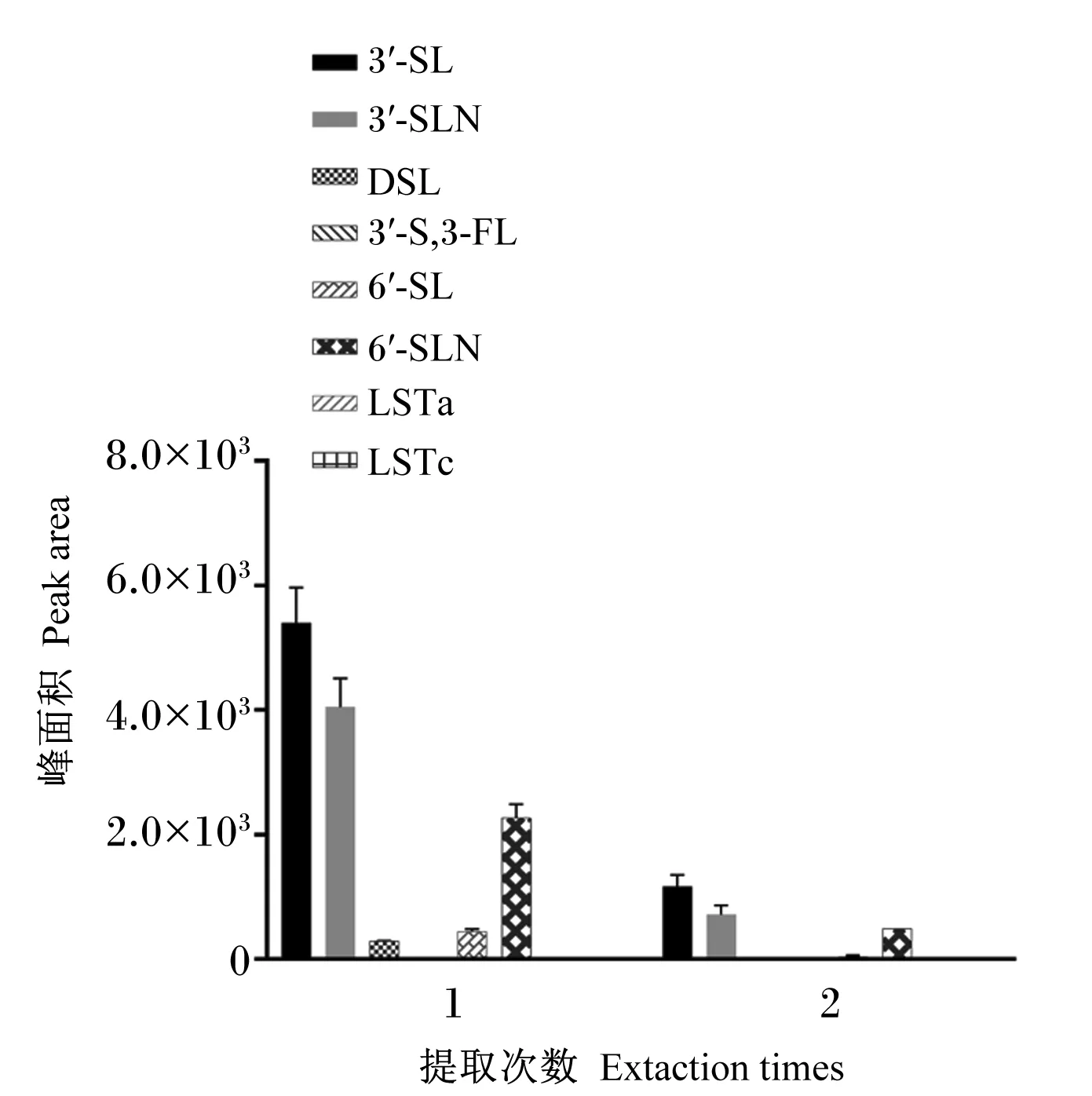
图2 提取次数对目标物峰面积的影响Fig.2 Effects of extraction times on peak area of target analytes
2.3 沉淀蛋白溶剂的筛选
由图3可知,利用乙腈作为沉淀蛋白的溶剂,各个目标化合物的峰面积均较甲醇的理想。因此,选用乙腈作为沉淀蛋白溶剂。
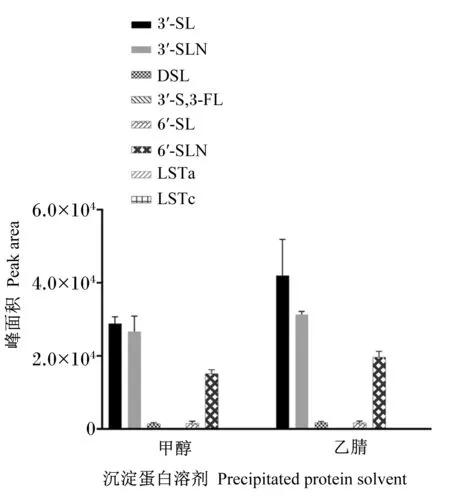
图3 沉淀蛋白溶剂对目标物峰面积的影响Fig.3 Effects of precipitated protein solvent on peak area of target analytes
2.4 沉淀蛋白溶剂比例的筛选
由图4可知,当样品与沉淀试剂的比例为1∶1和1∶2时,与其他比例相比,各个目标化合物的响应值相对较高。因此,为确保样品中的蛋白最大限度的沉淀,样品与沉淀试剂的比例选用1∶2。
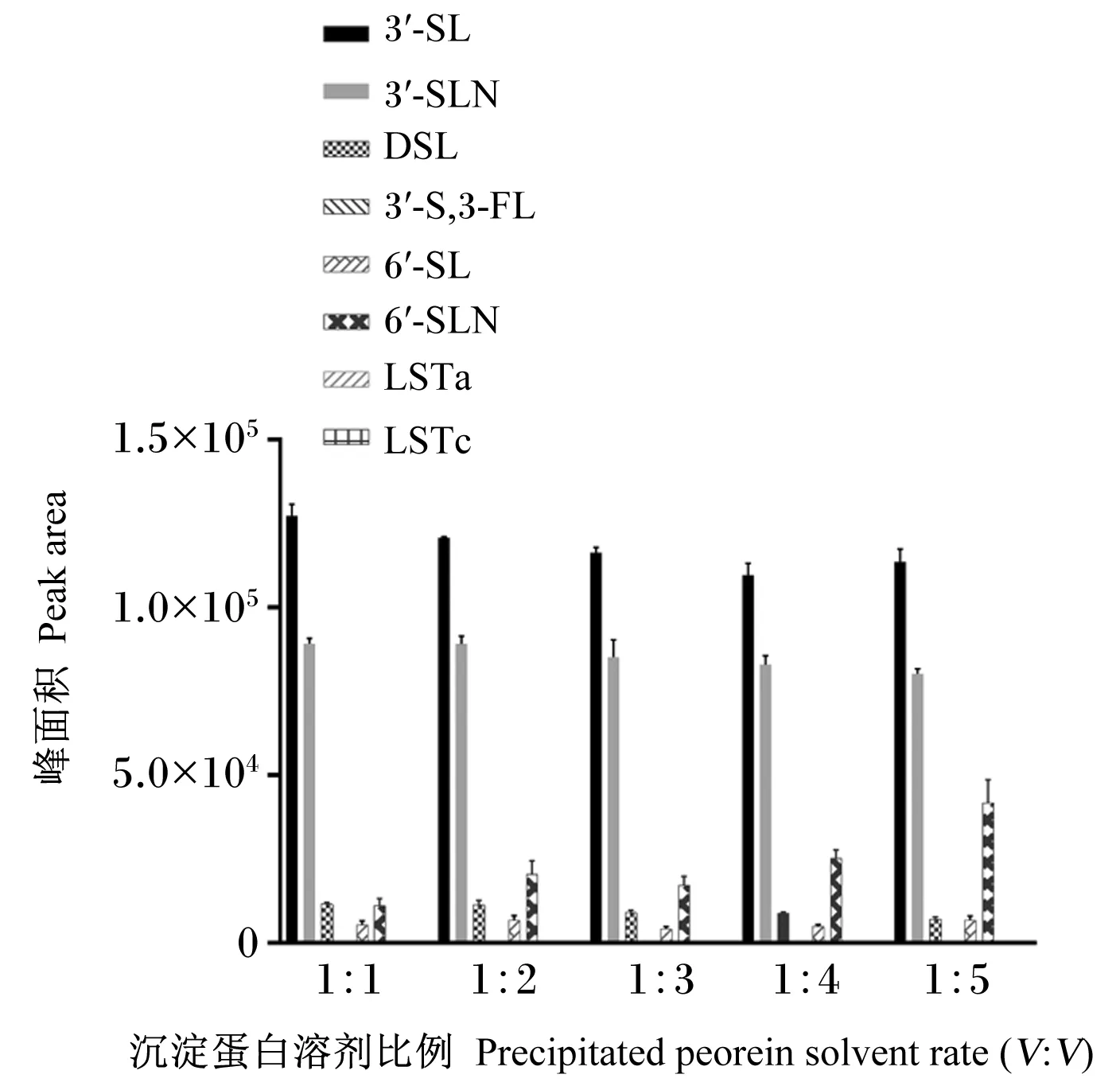
图4 沉淀蛋白溶剂比例对目标物峰面积的影响Fig.4 Effects of precipitated protein solvent rate on peak area of target analytes
2.5 沉淀蛋白时间的筛选
由图5可知,当沉淀蛋白时间在0.5~1.0 h时,各个目标化合物的响应值较为稳定,随着沉淀蛋白时间的延长,各个目标化合物的响应峰值略有下降,因此选择0.5 h作为沉淀蛋白时间。
2.6 氨基固相萃取小柱净化条件筛选
2.6.1 淋洗液的筛选
测定结果发现,乙腈∶水=70∶30和乙腈∶水=50∶50作为淋洗液,其中均有目标物3′-SL、6′-SL、6′-SLN和3′-SLN,用这2种淋洗液会造成目标物的损失,故不选用以上2种淋洗液淋洗。
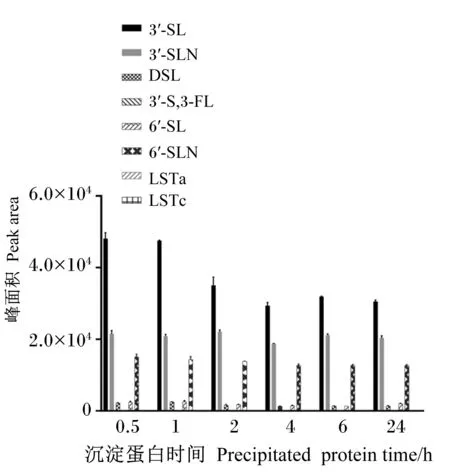
图5 沉淀蛋白时间对目标物峰面积的影响Fig.5 Effects of precipitated protein time on peak area of target analytes
2.6.2 洗脱溶剂的筛选
测定结果发现,当用洗脱液1或洗脱液2洗脱时,DSL检测到的量极少;当用洗脱液3洗脱时,8种目标物可以全部洗脱。因此选用15 mmol/L的乙酸铵水溶液(洗脱液3 500 μL)作为洗脱溶剂。
综上所述,筛选的前处理是以水为提取溶剂,样品(g)∶水(mL)为1∶25,以16 400×g,4 ℃离心10 min。超声提取2次,合并2次上清液;以乙腈沉淀蛋白,上清液与乙腈之间的体积比为1∶2,沉淀时间为0.5 h。再次离心后取上清液,用氨基柱(100 mg,1 mL)作为固相萃取柱,先用1 mL甲醇活化,取上清液500 μL上样,弃流出液;再用15 mmol/L的乙酸铵水溶液500 μL洗脱,洗脱液加入500 μL乙腈混合均匀,过0.22 μm滤膜,按照1.3的色谱和质谱条件上机测定。标准品和十二指肠内容物中目标化合物总离子流图和提取离子流图见图6。
2.7 标准工作曲线配制、线性范围及检出限
由表3可知,3′-SL、6′-SLN、LSTa、3′-S,3-FL、LSTc、3′-SLN、DSL和6′-SL的线性范围分别为5.00~2 000.00、2.50~100.00、9.59~94.00、10.0~100.00、27.80~200.00、4.00~80.00、6.00~120.00和16.10~642.00 ng/mL。8种目标物分别在各自的线性范围内线性关系良好,相关系数均达到0.991以上,检出限在0.335~8.330 ng/mL,结果见表3。
2.8 方法的精密性、重复性和回收率考察
由表4可知,方法精密性检测结果表明,各色谱峰的保留时间和峰面积的相对标准偏差分别不超过0.5%和5.0%,说明本方法精密性良好。方法的重复性检测结果表明,各色谱峰的保留时间和峰面积的相对标准偏差分别不超过0.5%和11.0%。回收率检测结果表明,样品加标平均回收率为77.3%~122.9%。
2.9 不同肠段样本中目标物的测定
选取羔羊空肠、回肠、结肠和盲肠4个不同肠段内容物样品,冷冻干燥研磨成粉后,分别称取0.025 0 g,按照优化的条件,测定不同肠段内容物中目标物的含量。由表5可知,4个肠段内容物中均未检出LSTc;不同肠段中检出目标物的含量不同,3′-SLN除在空肠中含量较低外,其余3个肠段中的含量接近。其他检出目标物的含量差异较大,空肠和回肠中的含量接近,盲肠中的含量除3′-SL高于结肠外,其他目标物的含量接近。
3 讨 论
方法建立在样品前处理条件优化过程中,开展了进样体积的筛选优化,分别考察了1和3 μL进样量的测定效果,由于样品中部分目标物质的浓度较低,目标物质的含量差异较大,在不污染离子源的前提下,尽可能的提高目标物质的响应值,因此进样体积确定为3 μL。
固相萃取小柱的筛选,基于肠道内容物提取液中可能含有的物质种类以及目标化合物的化学性质,分别采用磷脂小柱和氨基固相萃取柱对样品提取液进行净化。肠道内容物中含有部分脂质物质,有可能是主要杂质,因此优先选用磷脂小柱(30 mg,1 mL)作为固相萃取小柱进行净化。但是检测结果发现,目标物质全部被磷脂小柱截留,无法用乙腈或水等洗脱溶剂洗脱下来。选用氨基柱(100 mg,1 mL)作为固相萃取柱进行预试验,以混合标准溶液A作为样本,用水洗脱后,发现除DSL外,目标物响应值均与未过柱时接近,继续用15 mmol/L的乙酸铵水溶液洗脱,发现DSL响应值与未过柱时接近。因此,选用氨基固相萃取小柱可以净化提取液,降低杂质的干扰,从而获得更高的回收率及质谱检测灵敏度。
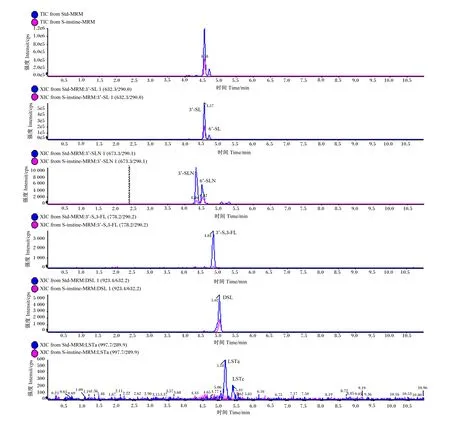
图6 标准品和十二指肠肠道内容物样本中目标化合物总离子流图和提取离子流图Fig.6 Total ion chromatogram and extracted ion chromatogram of target compounds in standard solution and duodenal intestinal content sample
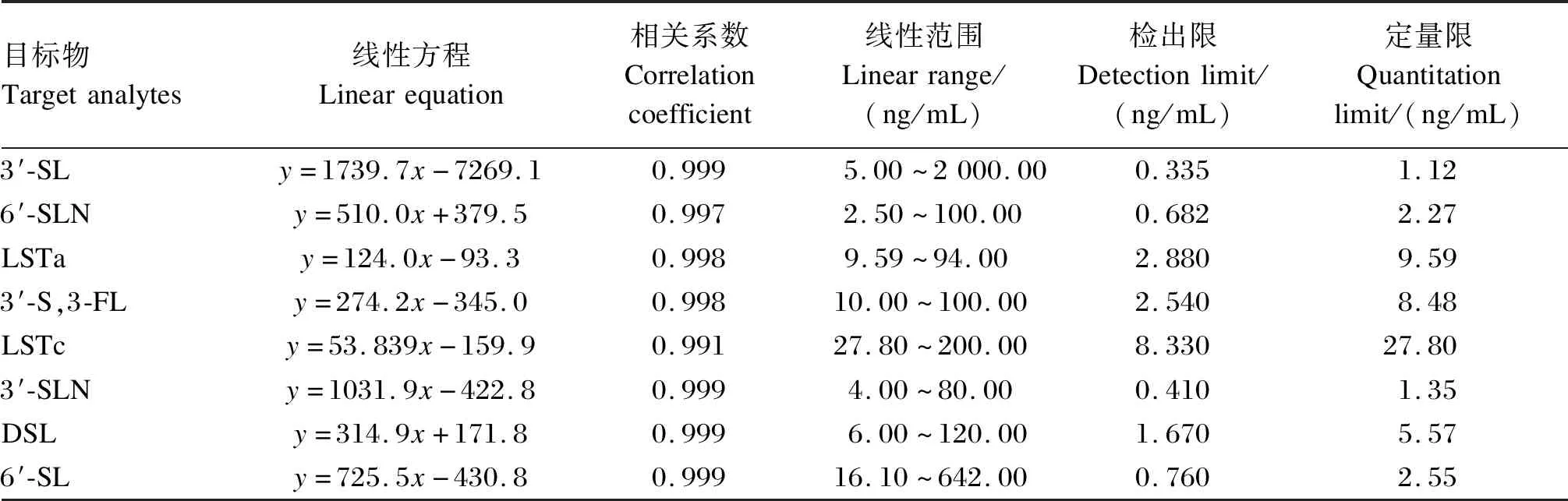
表3 8种目标物的线性范围、线性方程、相关系数、检出限以及定量限Table 3 Linear range, linear equation, correlation coefficient, detection limit and quantitation limit of eight target analytes
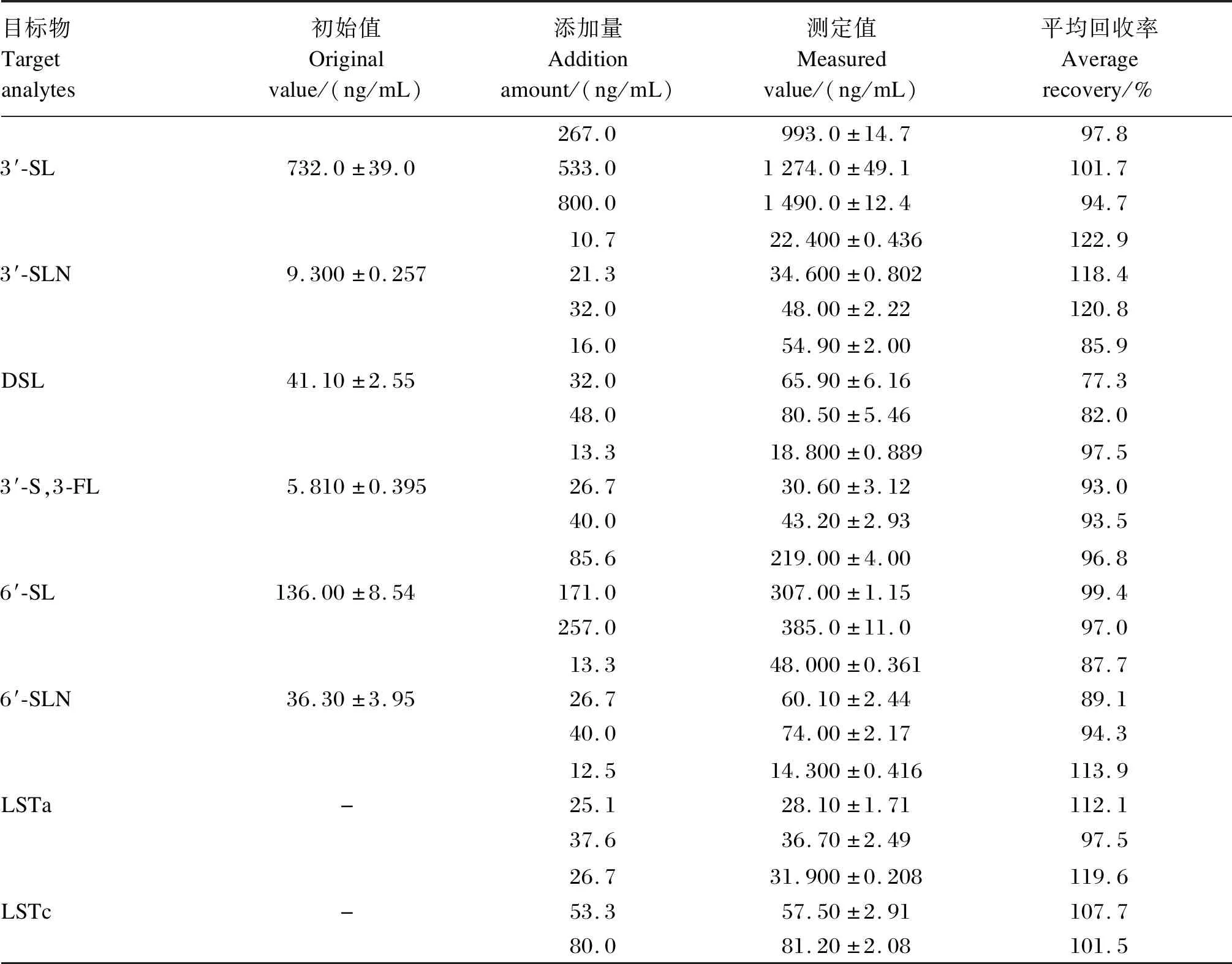
表4 样品加标回收率Table 4 Spiked recoveries of samples
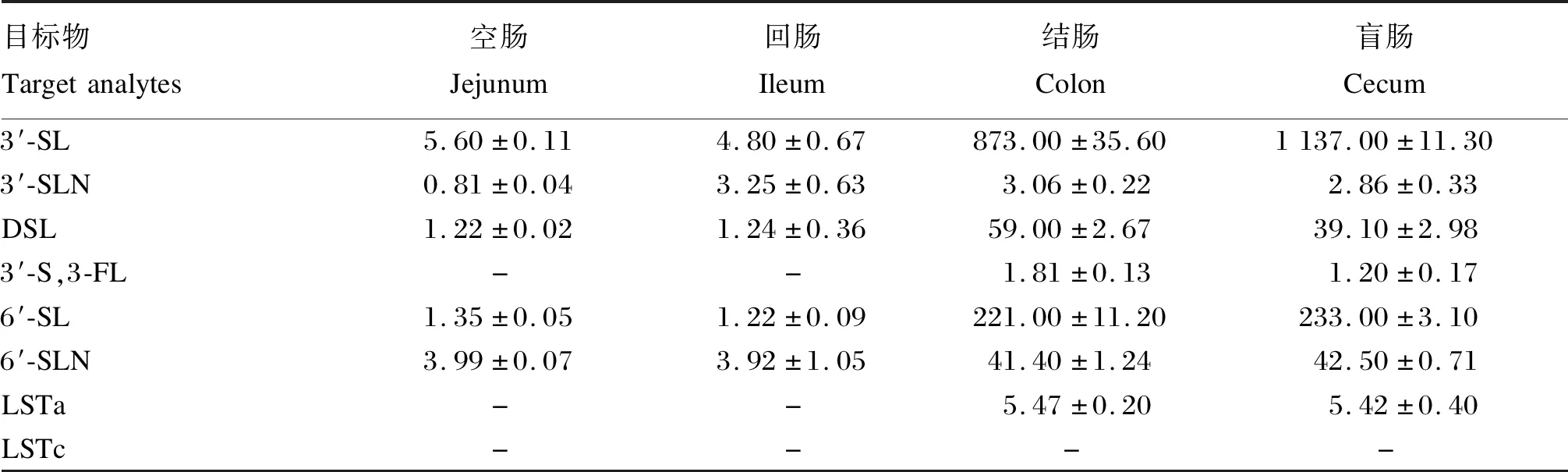
表5 不同肠段内容物样品中目标物测定结果Table 5 Results of target analytes in different intestinal content samples μg/g
本试验中通过单次进样,同时分析了8种酸性低聚糖,目标物在7 min内全部分离,与Zhang等[10]的方法相比,保留时间更短,显著提高了分析效率。Amanda等[11]建立的相关检测方法,未给出目标分析物的检出限,而本次建立的测定方法,明确了仪器的检出限,与前者的方法相比更加有利于样品的制备与分析。Ma等[12]建立了测定人乳中7种酸性低聚糖的高效液相色谱-质谱法,与前者相比,本试验建立的方法不仅检测的种类多,而且相同目标物的线性范围下限更低,表明该方法具有较高的灵敏度。由此可见,建立精准的测定分析酸性低聚糖在动物不同肠段含量以及变化规律的技术方法,可以更好地为深入系统研究动物肠道健康机制提供技术支撑。
4 结 论
本试验通过优化样品冷冻干燥、水提取、乙腈沉淀蛋白以及固相萃取等前处理,建立了测定羔羊肠道内容物中8种酸性低聚糖含量的液相色谱-质谱联用方法。8种组分的保留时间和峰面积的相对标准偏差分别小于0.5%和11.0%,线性相关系数均在0.991以上,检出限为0.335~8.330 ng/mL,样品平均回收率为77.3%~122.9%。该方法可为系统研究酸性低聚糖对动物肠道健康的作用机制提供技术支撑。
致谢:感谢中国科学院亚热带农业生态研究所公共技术中心(Institutional Center for Shared Technologies and Facilities of Institute of Subtropical Agriculture,CAS)对试验顺利开展提供的支持。
猜你喜欢
食品科学(2022年13期)2022-07-29
煤化工(2022年3期)2022-07-08
中山大学学报(自然科学版)(中英文)(2022年2期)2022-04-12
中国食品(2021年13期)2021-07-21
农产品加工(2020年20期)2020-11-14
农村百事通(2020年2期)2020-03-31
家庭百事通·健康一点通(2016年12期)2016-12-29
山东工业技术(2016年10期)2016-09-06
恋爱婚姻家庭·养生版(2016年5期)2016-05-06
饮食科学(2015年4期)2015-11-28
