
头孢菌素类药物中高分子杂质及检测技术进展
2022-11-08 08:51:46李伟
天津药学 2022年5期
李 伟
(天津市药品检验研究院,天津 300070)
头孢菌素类药物是β-内酰胺类抗生素中重要的组成部分,其分子构成中含有碳青霉烯结构,是β-内酰胺类抗生素中的7-氨基头孢烷酸(7-ACA)的衍生物。因其具有抗菌谱广、抗菌性能强、毒性低、耐酸耐酶性好以及分子结构易于改造等优点,自第一代青霉素被发现以来,发展迅速,现已研制出第四代头孢菌素,临床也被广泛使用。随着临床使用量的增加,有关此类药物不良反应的报道也日益增加[1-3]。常见的不良反应为速发型过敏反应,研究表明,速发型过敏反应是头孢菌素类药物中存在的高分子杂质引起的,而并不是人们通常认为的药物本身[4-6]。因此,考虑到头孢菌素类药物的临床用药安全性,对此类药物中的高分子杂质进行控制非常必要。近年来国内对包括头孢菌素类药物在内的β-内酰胺类抗生素中高分子聚合物的研究报道较多[7-11],但在头孢菌素类药物进行质量控制研究中,高分子杂质的控制仍然是其中的薄弱环节。本文总结了头孢菌素类药物中的高分子杂质来源、分类和形成机制、结构特性以及生理特性及致敏机制,并就包括葡聚糖凝胶G-10分子排阻色谱法、高效凝胶分子排阻法及反相高效液相色谱法等分离分析方法进行汇总,总结了《中国药典》中收载的关于此类药物中高分子杂质的分析方法,并对新技术的应用前景进行展望,为头孢菌素类药物中高分子杂质的研究及检测提供参考。
1 头孢菌素类药物中高分子杂质
1.1 高分子杂质的定义、来源及分类 头孢菌素类药物中的高分子杂质通常是指在生产、贮存、使用过程中产生的分子量大于药物本身的特定杂质的总称,其分子量一般大于1 000 Da[12]。
头孢菌素类药物中的高分子杂质按其来源划分,可分为外源性杂质和内源性杂质。外源性杂质一般针对全发酵类的抗生素(如青霉素类),主要包括发酵工艺中引入的多糖、多肽、蛋白等大分子杂质或者该类大分子与抗生素本身结合产生的大分子杂质。内源性杂质是头孢菌素自身聚合产生的寡聚物和多聚物的总称,其既可来源于生产过程,又可来源于运输贮存和使用过程[13]。随着生产工艺的改进和提高,以及对头孢菌素类抗生素结构的研究进一步明确,头孢菌素类抗生素已经不再依靠全发酵获得,而是通过在天然头孢菌素C的基础上对其结构改造得到半合成抗生素,引入外源性杂质的概率和含量都大大降低,因此对内源性杂质的控制则成为当前头孢菌素类抗生素中高分子杂质研究的重点。
1.2 高分子杂质的形成机制及结构特性 头孢菌素类药物中的高分子杂质的形成机制主要是聚合反应,早在1991年胡昌勤等[14]就对头孢菌素结构及其聚合反应的类型进行了研究。研究发现,聚合反应分为N型聚合反应和L型聚合反应。N型聚合反应:头孢菌素的母核为7-氨基头孢烷酸(7-ACA),如头孢噻啶、头孢呋新、头孢哌酮、头孢孟多、头孢噻酚等药物,由于在7-ACA母核的7位侧链中没有自由氨基等活性基团,此类型的聚合反应只依赖于头孢菌素的母核结构,7-ACA在特定条件下开环从而得到亲核的仲胺基,由于仲胺基具有较强的活性,与7-ACA另一侧的羰基亲核加成,形成自身聚合物。L型聚合反应:这种聚合反应由头孢菌素的侧链参与其中,7-ACA中的羰基碳原子受到7位侧链上存在的活性基团亲核攻击而形成聚合物。对于7位侧链中含有自由氨基等活性基团的化合物,聚合反应发生的情况又因受到pH值的影响而不同:当pH值小于7时,一般会在母核之间产生N型聚合反应,此时侧链不参与聚合;而当pH值大于7时,母核与7位侧链同时发生聚合反应,也就是N型和L型聚合反应都会发生,这种情况下产生的聚合物分子量也会比单纯的N型聚合反应下生成的聚合物分子量大,比如头孢拉定、头孢氨苄、头孢噻肟等。但是两种聚合反应的强度大小则与头孢菌素的化学结构本身有关。
1.3 高分子杂质的生理特性及致敏机制 过敏反应是抗原抗体相互作用产生的特异性反应,主要是由头孢菌素类药物在生产、贮存和配制过程以及进入人体后通过降解或者聚合产生的特定杂质(多为大分子杂质)与人体内的多糖、多肽及蛋白质等结合而引起的不良反应[15]。人体内的特异性抗体免疫球蛋白E(IgE)抗体与过敏反应密切相关[16]。头孢菌素类药物中7位侧链的结构是该类药物的抗原决定簇,其结构与青霉素6位侧链结构相似度越高,引发交叉过敏反应的概率就越大,反之,则不易引起交叉过敏反应[17]。胡昌勤等[18]利用离子对凝胶色谱法分离得到氨曲南中的大分子杂质,通过大鼠被动皮肤过敏反应(PCA)发现,Kav(有效分配系数)=0的高分子杂质能使PCA出现阳性反应(+),而Kav<0的氨曲南本身作对照,PCA则出现阴性反应(-)。后续研究[19]通过试验表明,头孢菌素类药物中的高分子杂质具有至少两个抗原决定簇,可作为多价半抗原引发IgE介导的Ⅰ型速发型特异过敏反应,而且聚合物的分子量越大,其抗原决定簇位点越多,引发过敏的可能性就越大。
2 高分子杂质的分析方法
2.1 葡聚糖凝胶G-10分子排阻色谱法 葡聚糖凝胶G-10分子排阻色谱法(Sephadex G-10 SEG)是凝胶色谱法(GPC)的一种,是高效分子排阻色谱法(HPSEC)的前身,该方法最早由我国的胡昌勤等[20]研究并推广使用。本方法的原理是利用高分子杂质与药物分子的分子量差异,通过凝胶色谱的分子筛作用,在流动相洗脱过程中,小分子量的药物分子保留在凝胶颗粒的空隙中,而大分子杂质由于体积大于凝胶颗粒之间的空隙而不能保留其中,最先被流动相洗脱进入紫外检测器中从而被检测出色谱峰,而药物分子及其降解产物等小分子化合物则晚于大分子杂质被洗脱出来。为了保证该方法的精密度、准确性和重复性,以浓度为1 mg/ml的蓝色葡聚糖2000溶液在流动相A[0.01 mol/L磷酸氢二钠溶液-0.01 mol/L磷酸二氢钠溶液(61∶39)]和流动相B(水)中保留时间、理论板数和拖尾因子来作为衡量标准,规定蓝色葡聚糖2000峰的保留时间在两种流动相系统中的比值应在0.93~1.07之间,对照溶液主峰和供试品溶液中聚合物峰与相应流动相中蓝色葡聚糖2000峰的保留时间比值均应在0.93~1.07之间。张红梅等[21]采用填充剂为葡聚糖凝胶G-10(40~120 μm)的玻璃柱(内径1.5 cm,柱长36.5 cm),对头孢孟多酯钠中的聚合物进行分离测定,结果表明,该方法能有效分离头孢孟多酯钠中的聚合物。该研究进一步考查了样品浓度对测定结果的影响,发现随着样品浓度的增加聚合物峰面积增大,但随之而来的弊端就是聚合物峰与药物本身峰的分离度越来越差,而样品浓度过低则聚合物峰面积过小,不易被检测出来,为了平衡峰面积与分离度之间的矛盾,最终确定样品的进样浓度为2.0 g/ml,在该浓度下,聚合物峰能与主峰达到有效分离,且峰面积适中。刘树彬等[22]建立了Sephadex G-10 SEG方法来测定注射用头孢地嗪钠中高聚物的含量,由于头孢地嗪对照品本身可以在水溶液中缔合,因此可以采用头孢地嗪作为自身对照。研究表明,该方法专属性强、分离效果好。由于头孢菌素类药物在水溶液中稳定性差,试验中考查了样品溶液的稳定性,分别取供试品溶液于0、1.5和3 h进样分析,聚合物含量分别为0.183%、0.240%和0.356%,表明供试品溶液中的聚合物含量随着放置时间的增加而增大,因此为了保证试验结果的准确性,要求供试品溶液在配制好以后立即进样测试。

表1 《中国药典》2020年版头孢菌素类药物聚合物的分析方法汇总(G-10法)
2.2 高效凝胶分子排阻法 高效凝胶分子排阻法(HPSEC)是在Sephadex G-10 SEG的基础上,采用更为高效的凝胶色谱填料作为填充剂,来实现对药物中存在的高分子杂质更好的分离效果,其分离机制仍是分子排阻。相较于Sephadex G-10 SEG法,HPSCE法不需要使用复杂的流动相系统,检验周期短,分离效果好,并能够在一定程度上实现分离不同分子量的高分子杂质。倪琼珠[24]采用填料为球状蛋白色谱用亲水硅胶(分子量范围1 000~10 000)的TSK gel-G2000SWXL高效凝胶色谱柱对盐酸头孢替安中的高分子杂质进行分析,在Sephadex G-10 SEG法使用的磷酸盐流动相A的基础上加入适当比例的乙腈,使磷酸盐缓冲液与乙腈的比例为90∶10,盐酸头孢替安中的高分子杂质能够与药物分子本身得到很好的分离,并且检出的高分子杂质含量比Sephadex G-10 SEG法高,检测时间15 min左右,该方法更高效简便。王娜等[25]在测定注射用盐酸头孢替安中的高分子杂质时,在系统适用性试验中增加了灵敏度溶液和系统适用性溶液。通过增加系统适用性溶液,可以更好的提升该方法对高分子杂质的检测能力。取供试品溶液于80℃加热2 min作为系统适用性溶液,通过加热破坏,产生更多的高分子杂质,在该色谱条件下,主峰前检出的8个高分子杂质峰均能与主峰达到良好的分离效果,且各杂质峰之间的分离度良好。通过该研究进一步说明,高效凝胶色谱法检测出的主峰前的高分子杂质峰不再像Sephadex G-10 SEG法那样将多种分子量大于药物本身的高分子杂质包含在一个色谱峰中,而是可以将不同分子量的高分子杂质进行有效的分离,这也为进一步研究高分子杂质的结构提供了有利的技术支撑。王小明等[26]在测定头孢克肟颗粒中聚合物含量时,采用分离效果更好的刚性、球形、亲水的多孔聚甲基丙烯酸酯树脂键合硅胶为填充剂的色谱柱(TSKgel G2500 PWxl,7.8 mm×30 cm,7 μm),分离出更多的高分子杂质。在该色谱系统中对头孢克肟特定杂质(杂质A、B、C、D、E、F)进行测定,发现杂质A、D、E在头孢克肟峰之前被洗脱出来,因此在计算头孢克肟高分子杂质时,将杂质A、D、E扣除,这样得出的聚合物含量结果更加准确,有效减少了在主峰前流出的已知特定杂质与辅料等共出峰的影响。崇小萌等[27]采用高效凝胶色谱法测定注射用头孢硫脒中的聚合物,并与Sephadex G-10 SEG法的测定结果进行比较,结果表明高效凝胶法能更有效的检测样品中的高分子杂质。杨倩等[28]通过优化流动相中磷酸盐缓冲液浓度、乙腈的添加比例、色谱柱温度等条件,建立了合理有效的方法,在主峰前检测出4个高分子杂质峰,该方法可以有效分离头孢替唑钠及其制剂中的高分子杂质。李敏等[29]建立的TSK凝胶色谱法,能够分离头孢妥仑匹酯片中高分子杂质,并有效去除辅料的干扰。熊雯等[30]利用两种凝胶色谱法测定注射用头孢地嗪钠中的高分子杂质含量,发现HPSEC法比Sephadex G-10 SEG法能更真实全面的反映出样品中的高分子杂质的情况。
HPSEC法与Sephadex G-10 SEG法相比,虽然能解决分离效果差、分析时间长、受辅料干扰严重、只能检测高分子杂质总量等诸多不足之处,但是其检验机制仍然是分子排阻,主峰前流出的色谱峰中不完全是高分子聚合物。由于高效凝胶填料在具备分子排阻作用外,还会产生非排阻作用,在疏水作用、吸附作用等干扰下,化合物的出峰顺序并不一定完全按分子量大小。崇小萌等[27]在测定注射用头孢硫脒中的聚合物的时候,通过柱切换联用质谱技术(LC-MS),对主峰前后的杂质峰进行结构确证发现,主峰后的3个杂质峰中,有2个色谱峰为高分子杂质峰。因此需要在此方法的基础上,联用LC-MS技术,对化合物结构进行分析,为后续的研究提供了新的思路。《中国药典》2020年版共收载头孢地嗪钠和头孢米诺钠两个头孢菌素类品种采用HPSEC测定样品中的高分子杂质,并将检验项目的名称定义为“有关物质Ⅱ”,这也进一步印证了,采用此方法检测出的主峰前的杂质不完全是高分子杂质,只是作为“有关物质Ⅰ”项下采用反相高效液相色谱法检测出的杂质的一个补充。分析方法汇总见表2。
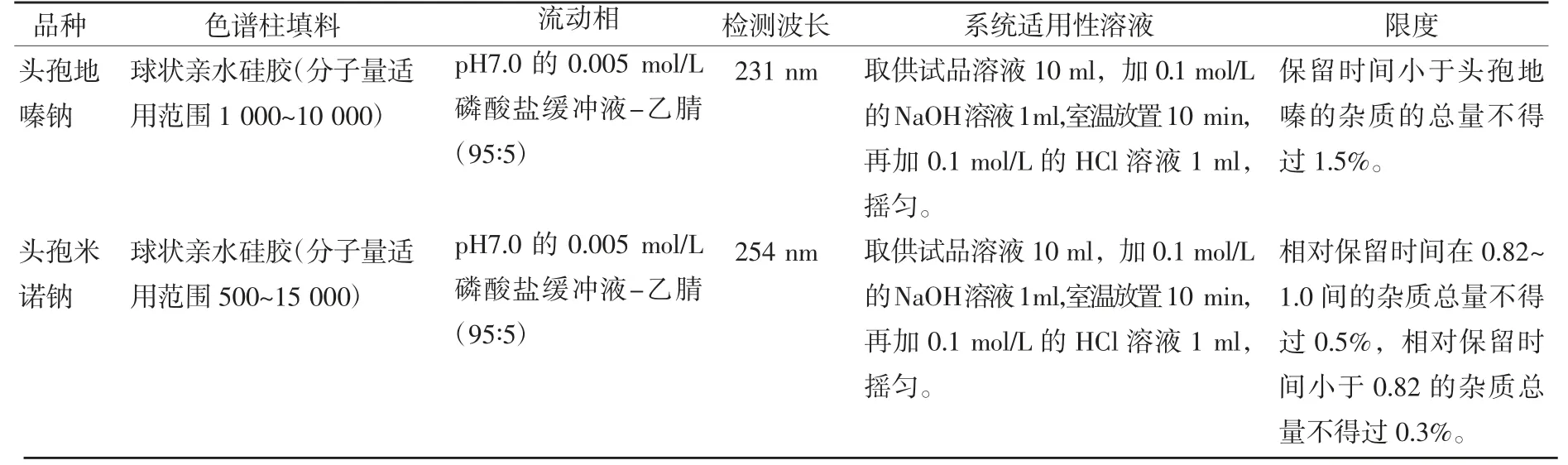
表2 《中国药典》2020年版头孢菌素类药物有关物质Ⅱ的分析方法汇总(HPSEC)
2.3 反相高效液相色谱法 反相高效液相色谱法(RP-HPLC)是在药物及杂质分离分析中最为常用的一种分析方法,样品中的主成分和杂质由于具有不同的极性大小,在不同的色谱柱填料、流动相的组成(pH值、缓冲盐浓度、有机相比例)、柱温、流速等参数共同作用下,不同极性的分子相继被洗脱出来,因此该方法具有更强的专属性和灵敏度。近年来,不少研究者利用RP-HPLC法来检测头孢菌素类药物中的高分子杂质[31]。唐浩等[32]采用RP-HPLC法同时测定头孢哌酮钠原料药中小分子杂质及指针性大分子杂质,通过优化流动相系统,增大有机相比例,并采用梯度洗脱的方法,在检测头孢哌酮已知杂质A、B、C、D、E、F的同时,在相对保留时间为1.5~2.5处发现两个未知杂质,采用LC-MS对未知杂质进行质谱分析,两个化合物的分子量分别为678 Da和791 Da,均大于头孢哌酮的分子量,可以作为头孢哌酮的高分子杂质进行控制。李进等[33]采用乙腈-甲醇水梯度洗脱的方法对头孢克肟中的杂质进行分离,在分离出的11个疑似高分子杂质中,经过LC-MS分析,确定其中5个为高分子杂质,其结构分别为头孢克肟二聚体异构体、头孢克肟脱水二聚体和头孢克肟二聚体以及头孢克肟二聚体衍生物,各个杂质分离度均符合要求,该方法专属性好。彭洁等[34]采用C8色谱柱同时测定头孢呋辛酯及其制剂中的小分子杂质及高分子杂质,通过改变流动相中磷酸盐的浓度及优化梯度洗脱程序,使高分子杂质分离良好,方法专属性强。
由于高分子杂质一般极性较弱,在RP-HPLC系统中出峰时间较晚,通常采用增大流动相中有机相比例,并同时使用梯度洗脱的方式来缩短检验周期。另外,利用RP-HPLC检测高分子杂质时,由于对高分子杂质较难定位,并且高分子杂质稳定性差,很难获得对照品,无法通过杂质对照品来定位。在方法开发过程中,通常要联用LC-MS等技术来对高分子杂质进行分子量确认和结构解析,为了提高新方法的开发效率、缩短研究周期、减少经费开支,研究者一般采用试验设计(DOE)来作为RP-HPLC方法开发的主要手段[35]。
3 结语与展望
头孢菌素类药物中的高分子杂质,尤其是内源性杂质是控制头孢菌素类药物质量的关键点,同时也是薄弱环节。随着科技水平的发展以及科研工作者对于高分子杂质控制的不断研究,人们对高分子杂质的结构特点、形成机制等有了深入的认识,已经从原来的总量控制发展到当前的对指针性化合物进行精准控制。利用更加先进的结构分析手段,比如质谱技术(LC-MS)[36]、核磁共振技术(NMR)[37]、LC/MS柱切换技术[38]、二维高效液相色谱法(2D-HPLC)[39]等对高分子化合物的结构进行分析,更好的控制药物中的高分子杂质含量。
1徐丽清,黄文璐.剖析第三代头孢菌素类抗菌药物药理作用及其临床合理用药情况[J].中国现代药物应用,2020,14(24):243-244.
猜你喜欢
功能高分子学报(2022年5期)2022-10-19 06:29:44
功能高分子学报(2022年5期)2022-10-19 06:29:44
功能高分子学报(2022年4期)2022-08-05 03:05:40
功能高分子学报(2022年4期)2022-08-05 03:05:40
纺织科学研究(2021年7期)2021-08-14 01:42:30
猪业科学(2018年5期)2018-07-17 05:56:20
纺织科技进展(2016年3期)2016-11-29 01:27:04
现代检验医学杂志(2016年1期)2016-11-12 13:19:54
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:21
国外医药(抗生素分册)(2016年1期)2016-07-10 12:02:35
