
泛素蛋白酶体抑制剂在急性髓系白血病中的研究进展
2022-11-03 02:37肖方楠张明英
中国医学科学院学报 2022年5期
肖方楠,张明英,周 圆
中国医学科学院 北京协和医学院 血液学研究所 血液病医院 实验血液学国家重点实验室国家血液系统疾病临床医学研究中心,天津 300020
急性髓系白血病(acute myeloid leukemia,AML)是一类起源于造血干/祖细胞的恶性克隆类疾病,具有高度异质性,其特点是白血病细胞异常增生、分化成熟障碍,并伴有凋亡减少。大多数AML患者初次诊断的年龄在60岁左右。传统AML治疗以化学药物联合治疗和造血干细胞移植为主要手段,对于60岁以上的老年患者,5年生存率只有10%~20%。同时,对于高危AML患者,复发率仍超过50%。虽然近年来靶向治疗、免疫治疗可以在一定程度上提高AML完全缓解率,但对于其早期复发率改善仍不明显。因此目前亟需开发有效且安全的新型治疗方法,以期一定程度上降低疾病复发率,从而实现AML患者的长期生存。
真核生物体内蛋白降解途径主要分为溶酶体途径、泛素化途径以及胱天蛋白酶途径。泛素蛋白酶体系统(ubiquitin-proteasome system,UPS)作为泛素化途径的重要成分,参与包括细胞凋亡、细胞周期、DNA修复和抗原呈递在内的多种生物学过程[2]。当UPS被抑制后,会激发多种信号通路有效杀伤细胞[3]。目前泛素蛋白酶体抑制剂在临床上主要用来治疗多发性骨髓瘤和套细胞淋巴瘤[4]。但已有数据显示,包括AML在内的其他多种类型肿瘤患者体内也可以检测到较高浓度的蛋白酶体以及泛素[5-6]。因此在单一治疗以及联合治疗中,泛素蛋白酶体抑制剂被认为是AML治疗的新策略[7-8],本文即针对其在AML中的研究进展进行相关总结与展望。
泛素蛋白酶体系统
2004年,以色列科学家Aaron Ciechanover、Avram Hershko和美国科学家Irwin Roseh因发现UPS调节的蛋白质降解获得诺贝尔化学奖。此后,诸多研究者发现UPS的失衡会导致多种疾病的发生。自从泛素蛋白酶体抑制剂硼替佐米于2003年被美国FDA批准应用于治疗套细胞淋巴瘤以及多发性骨髓瘤之后,越来越多的研究人员选择将UPS作为药物靶点进行相关研究。UPS通过选择性降解受损伤、错误折叠和短命蛋白以维持机体稳态。在该系统中需要降解的蛋白质首先经泛素化过程被标记,随后进入到26S蛋白酶体,经去折叠、去泛素化、肽酶剪切等步骤被降解成小分子氨基酸(图1)。
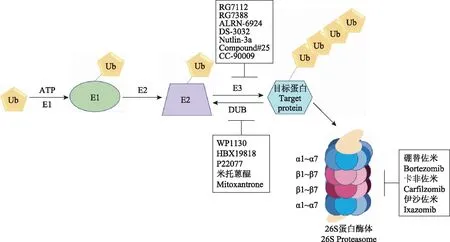
Ub:泛素;E1:泛素活化酶;E2:泛素结合酶;E3:泛素连接酶;DUB:去泛素化酶
Ub:ubiquitin;E1:ubiquitin-activating enzyme;E2:ubiquitin-conjugating enzyme;E3:ubiquitin-ligating enzyme;DUB:deubiquitinase
图1 泛素蛋白酶体系统及其小分子抑制剂概述
Fig 1 Overview of ubiquitin-proteasome system and its small molecule inhibitors
泛素化过程目标蛋白与泛素结合的过程由泛素活化酶(ubiquitin-activating enzyme,E1)、泛素结合酶(ubiquitin-conjugating enzyme,E2)以及泛素连接酶(ubiquitin ligase,E3)以ATP依赖的方式所催化,也可以被去泛素化酶(deubiquitinase,DUB)所逆转[2,9]。通过比较基因组分析,编码E1的基因很少,编码E2的基因有数十个,编码E3的基因达数百个,因此认为降解的靶向性主要是E3所介导[11]。目前研究显示人体内主要有600多个E3连接酶,根据结构域的不同,被分为3类:(1)RING(really interesting new gene)型E3连接酶,包括约600个成员,含量最丰富;(2)HECT (homologous to E6AP C terminus)型E3连接酶,约28个成员,可确定泛素化的特异性[15];(3)RBR (RING-between-RING) E3连接酶,18个成员,它们在UPS中起到至关重要的作用。
蛋白酶体最普遍的蛋白酶体形式是26S蛋白酶体,其作为主要的水解活性单位,由19S调节亚基以及20S催化亚基组成。19S调节亚基形似帽状结构,有6个ATPase和4个非ATPase亚单位,结合在20S催化亚基的1个或者2个末端,具有结合靶蛋白、去泛素化、展开标记蛋白的活性,同时还能以ATP依赖的方式将泛素化蛋白导入20S亚基中心活性位点室[18-20]。
20S催化亚基是一种高度保守的圆柱形复合体,由4个六角形环堆积而形成,入口处的2个外环由7个α单位组成,中间的内环由7个β单位组成。在真核生物体内,基于对特定氨基酸残基的识别裂解,蛋白酶体的活性被分为3大类:糜蛋白酶样、胰蛋白酶样和半胱天冬酶样,分别与β5、β2和β1亚单位相关[21]。
泛素蛋白酶体抑制剂在急性髓系白血病中的研究
迄今为止,泛素蛋白酶体抑制剂在临床主要用于治疗多发性骨髓瘤。但有研究表明,蛋白酶体的活性在AML患者血浆中异常升高[22]。关于白血病细胞系模型的一些研究表明,异常升高的蛋白酶体活性增强了白血病细胞的侵袭性[23],蛋白酶体抑制剂可诱导白血病细胞系的凋亡,显示出广泛的抗增殖和促凋亡活性[24-25]。因此,蛋白酶体抑制剂有望成为治疗AML的新策略。
20S蛋白酶体抑制剂硼替佐米(bortezomib)是第1个被FDA批准应用于临床的蛋白酶体抑制剂。在最近的研究中,硼替佐米被证实在体内使用可减弱白血病细胞蛋白酶体和NF-κB通路活性[26]。此外,硼替佐米还可通过控制FANCD2的单泛素化使肿瘤细胞对DNA损伤剂敏感,增强其他化疗药物的作用[27]。第2代蛋白酶体抑制剂卡非佐米(carfilzomib)近来也被证实能够有效抑AML中CD34+细胞增殖,具有比硼替佐米更强的细胞毒性[28],并且能增加化疗药物敏感性[29-30]。
然而,由于20S蛋白酶体受到抑制后,会使蛋白质泛素降解的途径被完全阻断,导致临床使用出现非靶效应。因此,亟需发现更具特异性的蛋白酶体抑制剂。
E3抑制剂临床数据显示,在白血病患者体内有着较高的泛素水平[22],提示通过抑制泛素化途径来治疗白血病可能是有效的。此外,E3决定了泛素化的特异性,因此抑制E3在癌症治疗中不良反应会更小,一些E3抑制剂已被广泛研究,例如靶向MDM2、SCFskp2的小分子化合物等。
MDM2与MDMX属于同源蛋白,二者均能够介导P53的泛素化降解。有报道证实在AML患者中出现MDMX过表达的情况[31],临床数据显示该比率可达到92%[32],因此抑制MDM2和MDMX以重新激活p53功能是治疗AML的一种有潜力的方法。最早研发的MDM2抑制剂为Nutlin-3a,它可以有效杀死过表达MDM2的野生型p53 ALL细胞,无遗传毒性,但其生物利用度较低,不太可能被用作单一疗法[10]。此后,优化的MDM2小分子抑制剂RG7112和RG7388等被投入临床试验中,并取得了较好的结果[33-34]。最近,Carvajal等[31]证实一种双重的MDMX/MDM2抑制剂ALRN-6924在白血病细胞中表现出强大的靶向活性。它能在白血病细胞系和原代AML患者细胞中诱导细胞凋亡和周期停滞,破坏白血病细胞克隆形成和持续增殖能力;在体内AML异种移植模型中,ALRN-6924也可抑制白血病的发生,提高存活率,且对白血病的治疗效果要强于MDM2单抑制剂RG7388。
SCF(Skp1-Cullin-Fbox)多亚基E3是RING E3中的一种亚型,参与调节细胞增殖和存活[2,35]。这种RING E3由接头(S-phase kinase-associated protein 1,SKP1)、支架(cullin 1,Cul1)、环指连接蛋白(RING finger)和识别蛋白质底物的F-box蛋白组成。S期激酶相关蛋白2(S-phase kinase-associated protein 2,Skp2)是研究最多的F-box蛋白之一,主要控制细胞周期调节因子如p21和p27的降解[36-37]。有实验证实一种Skp2激酶相关抑制剂C2能显著降低多发性骨髓瘤细胞的增殖,并诱导凋亡[38]。在髓系白血病中也可观察到Skp2水平的升高[39-40],有研究则证实Skp2促进了C/EBPα的蛋白酶体降解,而C/EBPα作为髓系分化的关键调节因子,其功能丧失即可导致分化受阻,诱导髓系白血病的发生[41-42]。Skp2缺失显著增加髓系细胞内源性C/EBPα水平,明显促进髓系细胞的分化,抑制其集落形成能力,表明Skp2有可能成为治疗分化受阻的髓系白血病的潜在靶点[41]。但有关S期激酶相关蛋白抑制剂在AML中的研究仍旧较少,其在髓系白血病细胞中的药理学作用有待进一步研究。
FBW7作为SCF型泛素连接酶复合物的重要组成部分,能控制许多蛋白的泛素化以及降解,如c-Myc、细胞周期蛋白E(Cyclin E)等[43]。Zhu等[44]利用Oncomine的数据库分析FBW7在不同肿瘤和正常组织中的mRNA水平,发现FBW7在白血病中的表达较高,FBW7的致癌作用在多发性骨髓瘤等血液肿瘤中也被得到证实。随后有研究表明在AML细胞中抑制FBW7可恢复粒细胞集落刺激因子信号,从而增强信号转导与转录激活因子3(signal transducer and activator of transcription 3,STAT3)活性,促进粒细胞分化,且FBW7与众多肿瘤发生相关通路如p53、NF-κB等具有相关联[43,45]。因此,靶向FBW7也可能为AML患者提供一种新的治疗思路。
除E3抑制剂可有效治疗肿瘤外,目前有研究认为E3调节剂在这方面也有一定的作用,例如CC-90009(新型E3调节剂),作用于E3复合物CRL4CRBN(CUL4-DDB1-CRBN-RBX1),靶向针对GSPT1进行泛素化和蛋白酶体降解,从而耗尽GSPT1蛋白迅速诱导AML细胞凋亡,减少原发AML患者的白血病植入和白血病干细胞数量[46]。
DUB抑制剂蛋白质泛素化可被DUB所逆转,目前已经发现有约100种DUB,可被分为6个家族,其中大多数属于半胱氨酸酶[47]。DUBs执行3个主要功能:形成成熟形式的泛素,去除修饰蛋白质上的多泛素链以及剪切泛素链、编辑泛素化修饰[35,47-48]。
USP9X是一种可以切割多种泛素链的DUB。目前已经观察到,在慢性粒细胞白血病中USP9X的表达增加,并与抗凋亡蛋白MCL1的表达增加相关[49]。WP1130作为一种部分选择性DUB抑制剂,被证明可抑制USP9X,阻断慢粒细胞中异常激活的异常BCR/ABL激酶,诱导细胞凋亡[13]。近来的研究中,这种DUB抑制剂又被证实能诱导FLT3-ITD的聚集体易位,从而下调FLT3-ITD,并导致氧化应激协同诱导AML细胞凋亡[50]。USP10的抑制剂HBX19818和P22077也被证实具有类似的生物效应[16]。Yang等[51]最近证实USP10抑制剂能有效破坏脾酪氨酸激酶(spleen tyrosine kinase,SYK)驱动的白血病细胞增殖,并诱导SYK蛋白降解,为治疗酪氨酸激酶抑制剂(tyrosine kinase inhibitor,TKI)耐药患者提供一种新思路。
此外,USP7作为新的治疗靶点开始引起人们越来越多的关注。USP7最初通过大规模RNAi筛选而被认为是有效的抗癌靶点[52]。在正常情况下USP7优先与p53的负调控因子HDM2结合,维持体内低p53水平。当DNA受到损伤后,USP7发生去磷酸化,降低对HDM2的亲和力,促进与p53结合,稳定p53蛋白,启动p53依赖DNA损伤反应,有利于DNA损伤修复,在DNA复制过程中也被证明可以有效维护染色体稳定,促进复制叉的进展[53]。Cartel等[54]最近进一步证明,USP7抑制能够以非p53依赖的方式有效抑制体内外AML细胞的增殖,与化疗药物阿糖胞苷具有协同作用,还提供了USP7是耐药AML细胞的重要组成部分的证据[54]。并且有相关研究证实DUB小分子抑制剂PR619对AML细胞系具有独立于DUB抑制活性的DNA拓扑异构酶Ⅱ毒性[55]。以上实验均提示DUB抑制剂在AML中具有极大的治疗潜力。
总结和展望
泛素蛋白酶体抑制剂在多发性骨髓瘤上的成功应用提示UPS在癌症发生发展过程中的重要性,许多与此相关的研究尚在进行中。AML作为一种生物学和临床上的异质性疾病,虽然对其进行风险分层优化了传统治疗方案,但患者的长期生存依然较差,对于难治、复发患者难以进行有效治疗。因此在AML疗法的研发中,期待新疗法的出现。目前,针对UPS的各种成分的小分子抑制剂被证实在治疗AML方面具有较为显著的效果,临床上相关数据也佐证了这一点(表1)。然而,仅有小部分UPS抑制剂被真正的应用到临床实践上,靶向性、耐药性以及有限的疗效限制了它们的实际应用,如果可以克服这些问题,UPS抑制剂将可能成为治疗AML的新兴方法。

表1 泛素蛋白酶体抑制剂在临床中的应用Table 1 Clinical application of ubiquitin-proteasome system inhibitors

续表1
猜你喜欢
医学综述(2020年11期)2020-02-16
华东师范大学学报(自然科学版)(2018年2期)2018-05-14
科学中国人(2017年36期)2017-06-09
现代检验医学杂志(2016年5期)2016-08-20
中华老年多器官疾病杂志(2016年9期)2016-04-28
西南军医(2016年4期)2016-01-23
中国继续医学教育(2015年4期)2016-01-07
中国病理生理杂志(2015年8期)2015-12-21
浙江大学学报(农业与生命科学版)(2015年4期)2015-12-15
中国医学科学院学报(2015年5期)2015-03-01
