
基于SSR标记的我国主栽日本栗品种(系)遗传结构分析和指纹图谱构建
2022-10-29 02:56聂兴华王碧瑶练蔓青郑瑞杰
核农学报 2022年11期
聂兴华 刘 松 王碧瑶 李 琰 练蔓青 秦 岭,2 郑瑞杰 邢 宇,2,*
(1 北京农学院植物科学技术学院,北京 102206;2 林木分子设计育种高精尖创新中心,北京 102206;3 辽宁省经济林研究所,辽宁 大连 116000)
栗营养含量高,脂肪含量低,富含核黄素和多种维生素,具有良好的可食用价值和药用价值,是世界上重要的干果作物[1-2]。现今公认的栗属植物共有7个种,分别为锥栗[Castaneahenryi(Skam)Rehd.et Wils.]、板栗(CastaneamollissimaBl.)、茅栗(CastaneaseguiniiDode)、欧洲栗(CastaneasativaMill.)、日本栗(CastaneacrenataS.et Z.)、美洲栗[Castaneadentata(Marsh.)Brokh.]和美洲榛果栗(CastaneapumilaMill.)[3-4]。日本栗,又称丹东栗,主要分布于日本、韩国、朝鲜以及我国辽宁和山东地区。我国现今分布和栽培的日本栗有两个分支,其一为20世纪20年代由朝鲜传入山东适应暖湿环境的文登栗,其二为位于丹东市和辽南地区适应干旱寒冷环境的丹东栗[3]。日本栗不仅具有坚果大、适应性强、耐寒、丰产性高等优点,而且对栗疫病、墨水病等病害表现出较好的抗性,是世界各地栗属植物获取抗性基因的重要亲本。现阶段育种者已培育出诸多抗性强、丰产性高的欧日、中日杂交品种[5-8]。近些年,随着日本栗良种在我国东北部省区的不断推广,其经济效益与生态效益得到显著提升,已发展成我国辽宁省山区的主要支柱产业[9]。中国板栗和日本栗在历史上有着相互引种且部分生态区域重合,种间渐渗类型较多,特别是日本栗品种选育谱系分类不清,加上各产区使用没有标签的接穗进行繁殖,导致日本栗品种资源混乱的现象,资源鉴定的研究较为滞后。因此,探究科学有效的分子鉴定方法,对日本栗品种(系)资源保护利用具有重要意义。
上世纪80年代以来,随着分子标记技术不断发展,该技术被广泛应用于植物的遗传多样性、遗传图谱、指纹图谱和数量性状位点(quantitative trait locus, QTL)分析[10-13],其中简单重复序列(simple sequence repeat,SSR)具有共显性、稳定性、可重复性和多态性等特点,是诸多分子标记中应用最为广泛的标记之一。在日本栗的最新研究中,SSR主要用于遗传多样性和性状关联分析等领域。2018年Nishio等[14]利用SSR和单核苷酸多态性(single nucleotide polymorphism, SNP)对99份日本栗资源进行关联分析,挖掘了12个与坚果性状相关的分子标记。2020年Nishio等[15]使用36个SSR分子标记对230份日本栗品种资源和中国板栗品种资源进行遗传多样性评估和亲缘关系分析,阐明了日本栗和中国板栗品种之间具有很高杂交育种前景。2021年Terakami等[16]利用374个SSR分子标记构建了日本栗的遗传图谱,并定位到与垂枝性状相关的标记位点。日本栗虽然在世界栗属种质资源中发挥着非常重要的作用,但是目前有关日本栗与其他栗属植物的亲缘关系和日本栗主栽品种分子鉴定的研究却鲜有报道。基于此,本研究全面收集我国主栽日本栗品种,并利用荧光SSR分子标记对124份栗属植物资源进行亲缘关系分析,解析日本栗的分类地位和日本栗品种的遗传结构,旨在为日本栗在栗属植物中的分类地位和应用价值提供支撑;并构建59份日本栗主栽品种的指纹图谱,以期为日本栗在繁殖过程中品种的纯度和品种的独特性提供基础数据,为今后日本栗的资源的鉴定和种质创新提供参考,同时为日本栗品种知识产权的保护提供理论基础。
1 材料与方法
1.1 试验材料
本研究的供试材料采于2019—2021年,共计124份,详细信息见表1,包括30份板栗资源、13份锥栗资源、22份茅栗资源和59份日本栗品种(系)资源,其中日本栗采自辽宁经济林研究所日本栗资源圃。每份供试材料采集嫩叶6片,液氮冷冻后,-20℃条件保存于低温冰箱。
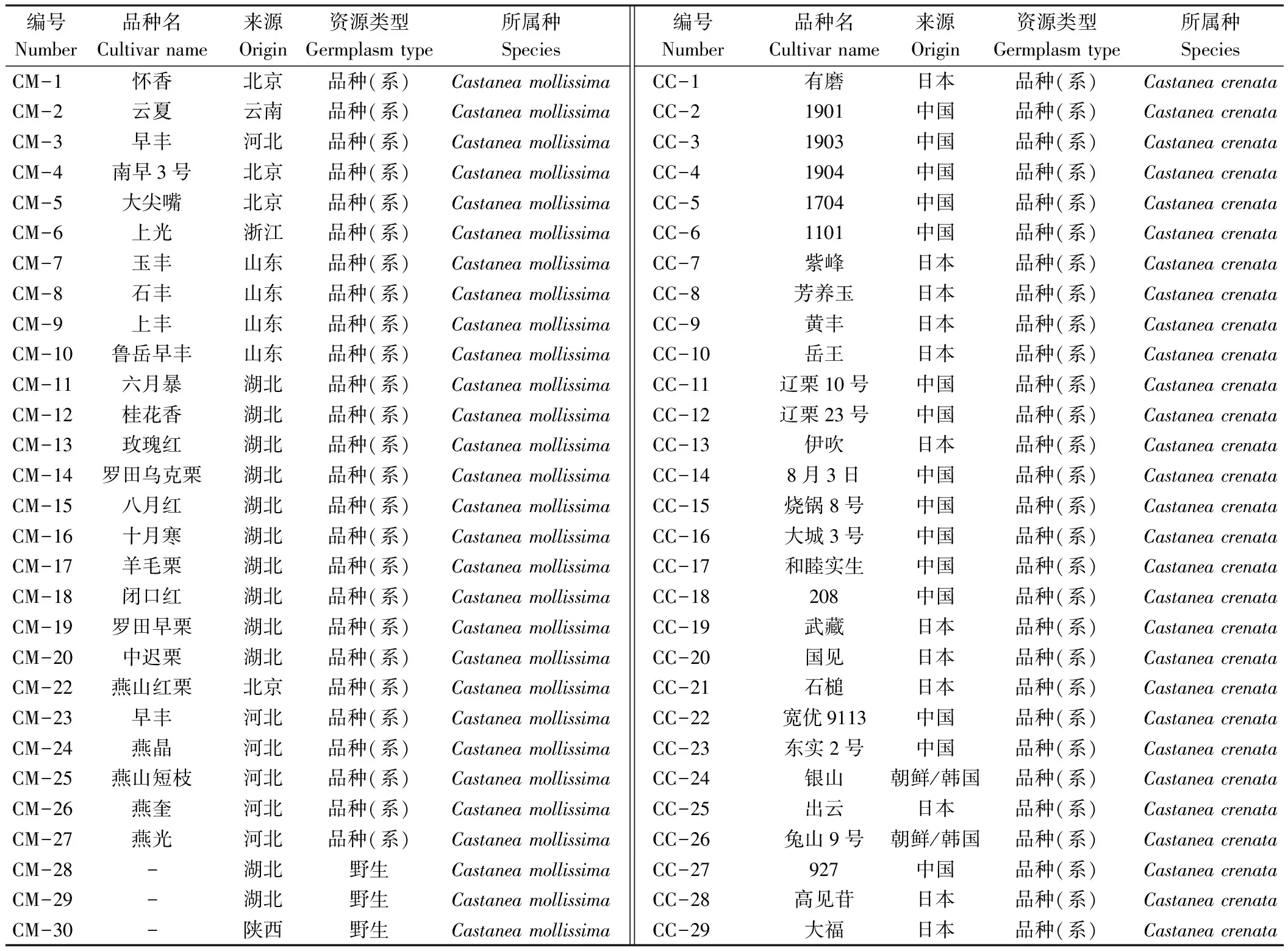
表1 124份供试材料信息表
1.2 试验方法
1.2.1 DNA提取 板栗基因组DNA提取采用改良的十六烷基三甲基溴化铵(cetyltrimethylammonium bromide, CTAB)方法[17]。提取DNA后,使用1%琼脂糖凝胶电泳和NanoDrop One分光光度计(Thermo Fisher Scientific, 美国)分别检测DNA的质量和浓度,并将每份DNA的浓度稀释至20~50 ng·μL-1以备扩增使用。
1.2.2 SSR分子标记的来源 本试验使用的17对SSR引物由北京农学院板栗分子发育生物学实验室前期筛选所得[17],详细信息见表2。引物由北京擎科生物科技有限公司合成。

表2 17对SSR引物的信息
1.2.3 毛细管电泳SSR-PCR扩增体系 扩增体系20 μL:1 μL DNA底物、10 μL 2×Taq Plus PCR MasterMix(Biomed, 北京)、10 μmol·L-1正向引物(0.1 μL)、10 μmol·L-1反向引物(0.3 μL)、10 μmol·L-1含荧光标记(FAM,HEX,ROX或TAMRA)的M13引物(0.2 μL)(擎科,天津)和8.4 μL ddH2O。使用T100 PCR仪(Bio-Rad, 美国)进行扩增。PCR反应程序:94℃预变性3 min;94℃变性30 s,56℃退火 30 s,72℃延伸30 s,36个循环;72℃延伸10 min。10℃条件下保存。
1.2.4 毛细管电泳位点信息检测 将不同荧光的PCR扩增产物混合在一起,然后使用ABI 3730XL DNA测序仪(Applied Biosystems,美国)进行位点信息检测,再使用Gene Marker v 2.2.0软件(Soft Genetics LLC,美国)精准读取片段大小。
1.3 数据分析
使用PowerMarker 3.25[18]和GenAlEx 6.51[19]软件进行计算,包括多态信息含量(polymorphism information content, PIC)、主要等位位点频率(major allele frequency, MAF)、等位位点数量(number of alleles,Na)、遗传多样性(genetic diversity,GD)、期望杂合度(expected heterozygosity,He)、鉴别概率(probability of identification,PI)等遗传多样性指标。使用PowerMarker 3.25评估各资源间的Nei’s遗传距离,并使用FigTree v1.4.3构建UPGMA聚类树。使用Structure 2.3.3[20]的贝叶斯模型进行群体结构的分析,参数迭代次数(length of burnin period)设置为 10 000,马尔可夫链重复次数(number of MCMC Reps after burnin)设置为100 000。使用GenAlEx 6.51对供试资源进行主坐标分析(principal co-ordinates analysis, PCoA)。使用Prism 9.0进行指纹图谱的图像化处理。
2 结果与分析
2.1 日本栗的亲缘关系与聚类特点
为了了解日本栗在东亚栗属的分类地位及其与其他栗属植物的亲缘关系,本试验加入了13份锥栗、22份茅栗和30份板栗资源进行聚类和群体结构分析。结果显示(图1-A、C),整个东亚栗属植物为单系群,存在明显的种间界限。锥栗位于所有资源的基部位置,板栗位于次基部位置,随后日本栗和茅栗组成了一个姊妹系。说明日本栗与茅栗具有较近的亲缘关系。同时在聚类分析中发现,板栗资源CM-18聚于茅栗资源,结合群体结构分析结果显示,CM-18资源中11.8%遗传成分来之茅栗,判定其为种间杂种。
日本栗品种资源单独聚类结果显示(图1-D),所有资源主要分为2个分支,其中资源CC-12(辽栗23号)、CC-14(08-3)、CC-18(208)、CC-22(宽优9113)、CC-27(927)、CC-43(辽丹58)、CC-44(9912)、CC-45(辽栗15号)、CC-55(包营独果)和CC-58(辽丹61)等10份来源于我国的日本品种(系)资源组成了簇1(Cluster 1);其余日本栗品种资源独立一支组成簇2(Cluster 2),包括25个日本本土品种资源、8个朝鲜半岛品种资源和16个中国品种资源且均为已知中日杂交品种(系),其中5个朝鲜半岛品种位于Cluster 2的基部位置。
注:A:124份栗属植物的系统发育树;青色部分为日本栗,紫色为茅栗,粉色为板栗,绿色为锥栗;B:不同K值的ΔK分布;C:124份供试材料的群体结构;D:日本栗品种的种内系统发育树;蓝色部分为Cluster 1,红色部分为Cluster 2。
2.2 日本栗品种资源的遗传多样性
对59个日本栗品种进行荧光毛细管电泳检测,在17对SSR引物中共获得131个等位位点(表3)。每个基因座上的等位位点(Na)为5~13,平均值7.706个;主效等位位点频率(MAF)为0.194~0.732,平均值为0.442;观察杂合度(Ho)为0.068~0.831,平均值为0.568;期望杂合度(He)为0.370~0.875,平均值为0.689;多态信息含量(PIC)为0.373~0.815,平均值约为0.605;香农指数(I)为0.733~2.233,平均值为1.459。以上数据表明,选用的SSR标记具有丰富的多态性,供试日本栗品种(系)资源具有较高杂合度且遗传多样性丰富。
2.3 日本栗品种资源的群体结构与主坐标分析
为了进一步评估日本栗品种资源的遗传特征,本试验利用structure 2.3.3和GenAlEx 6.51进行了群体结构和主坐标分析(图2)。结果显示,当K=2时,ΔK有最大值,表明59份日本栗品种(系)资源最佳可为两个类群。大部分资源为混合类型,说明两个分群之间存在着广泛的杂交现象,其中组1中Q值大于0.8的资源分别是CC-12(辽栗23号)、CC-14(08-3)、CC-18(208)、CC-22(宽优9113)、CC-27(927)、CC-34(山大)、CC-35(1401)、CC-43(辽丹58号)、CC-44(9912)、CC-45(辽栗15号)、CC-55(包营独果)和CC-58(辽丹6号),这些品种与聚类树Cluster 1的资源一致,分别是中国境内的日本栗品种(系),同时也与主坐标分析中组1聚类资源一致。组2 Q值大于0.8的资源共13个,分别是CC-1(有磨)、CC-3(1903)、CC-7(紫峰)、CC-8(芳养玉)、CC-11(辽栗10号)、CC-13(伊吹)、CC-19(武藏)、CC-20(国见)、CC-21(石槌)、CC-25(出云)、CC-26(兔山9号)、CC-31(大丹波)和CC-32(1703),其中日本本土品种9个,中国品种3个、朝鲜半岛品种1个。当K=3时,CC-8(芳养玉)、CC-25(出云)、CC-53(大国)、CC-3(1903)、CC-37(玉光)、CC-49(农林8号)、CC-9(黄丰)和CC-15(烧锅8号)等组成了新的分群。综上可知,中国境内的日本栗品种与日本本土的日本栗品种存在一定程度的分化,且各资源间存在着频繁的基因交流。

注:A:利用structure 2.3.3计算不同K值的ΔK分布,K代表样品分群个数,ΔK值越大支持所对应的K值越合理;B:品种资源的群体结构;C:品种资源的主坐标分析(PCoA)。红色圈为部分中国境内的资源,蓝色圈主要是日本和朝鲜半岛的资源,其中绿色主要是日本本土的资源。
2.4 日本栗品种的指纹图谱构建
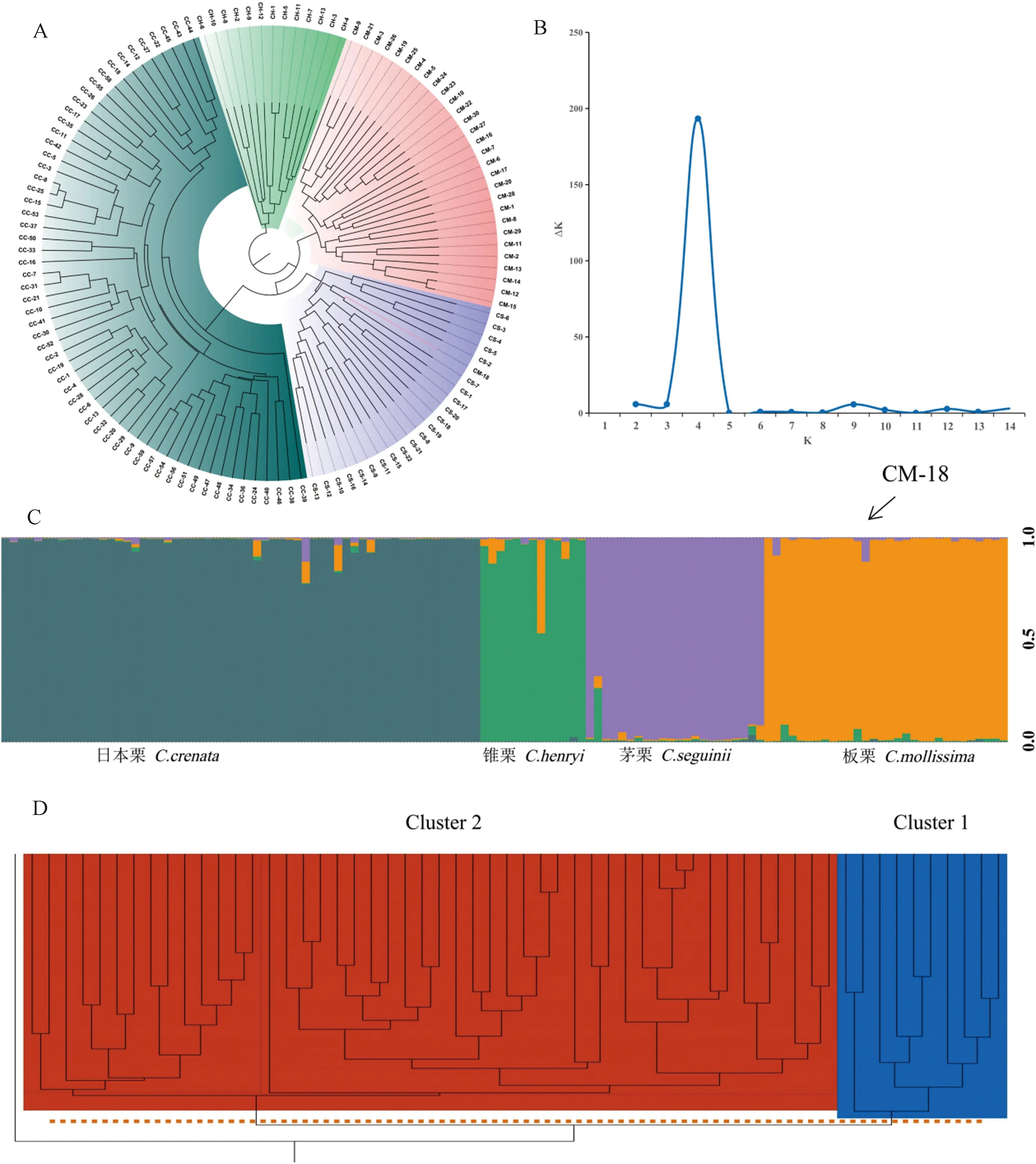
通过17个SSR分子标记的位点检测,共获得多态性和可重复等位位点组成的232个基因型。基于基因分型数据在GenAlex 6.51中对59个日本栗品种进行了多基因座匹配分析,未检测到基因型完全一致的两个品种,表明在59个日本栗品种都有其独特的SSR多位点基因型组合。为了评估17个分子标记的指纹识别能力,计算了两个关键统计值PI和PIsibs(表3)。结果表明,每个引物的PI值范围为0.028(CmSI0922)~0.424(CmSI0614),平均值为0.151。假设所有标记位点独立分离,两个随机个体在所有17个引物中存在完全相同多位点基因型的概率估算为3.471×10-16。PIsibs被定义为PI上限,17个SSR标记的PIsibs在0.319(CmSI0922)~0.574(CmSI0396和CmSI0561)之间,平均每个位点为0.443,组合的PIsibs为7.764×10-7。

表3 17对SSR分子标记在59个日本栗资源的遗传数据
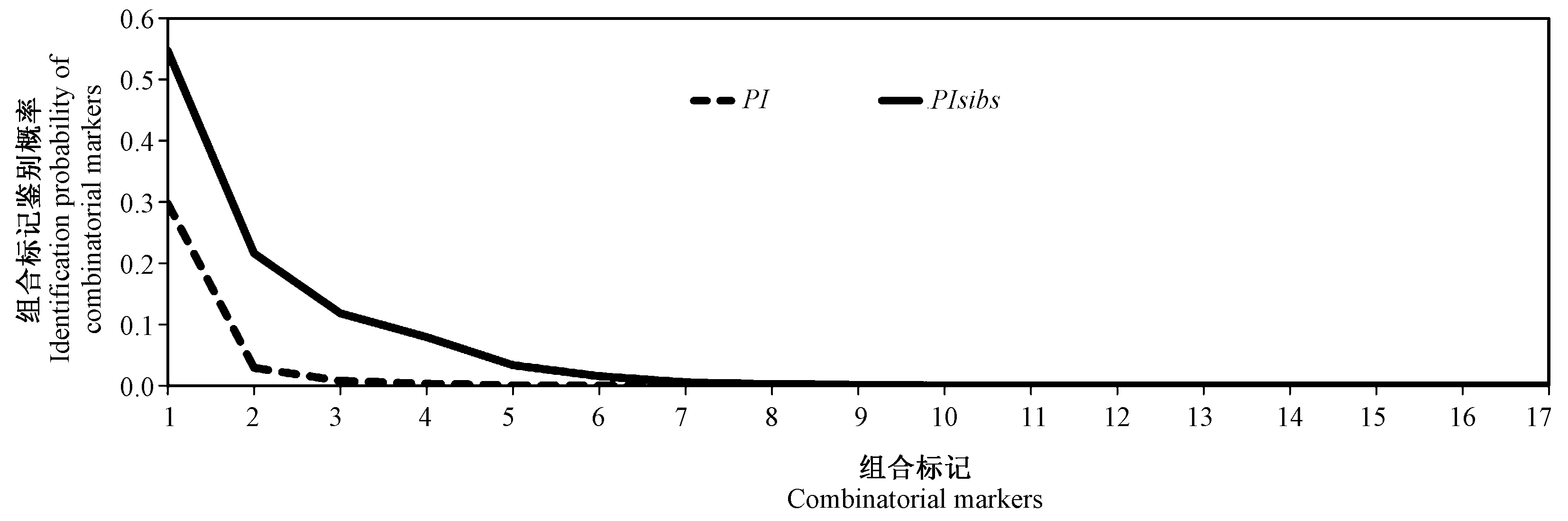
根据位点组合数据,计算了17个SSR标记在不同标记数量随机组合下的识别能力,以评估能完全区分所收集的日本栗品种需要的标记数量。结果显示,当3个SSR标记相结合时,PI值接近0,说明在标记数为3时,所有收集日本栗品种可以被独立鉴定(图3)。根据核心标记选择原则和基因型数据结果,本试验筛选出3个SSR分子标记作为核心标记。首先选择具有较高PIC值和较低PI值的CmSI0922,该引物将59个品种独立分为32组,从所有品种中独立分离出14个品种。当添加第二个标记(CmSI0702)进行进一步分析时,可以唯一识别59个品种中的48个,占所有品种资源的81.36%。添加第三个(CmSI0658)标记进行分析时可以完全区分59个日本栗品种,而3个核心标记的PI和PIsibs组合分别为2.256×10-4和5.184×10-2,随后总结了这些核心标记的等位基因大小和频率(图4)。由此,本研究使用这3个核心标记以热图的形式构建了59个日本栗品种的指纹图谱(图5),其中每个标记有两个等位位点组成,每列中的一个颜色代表一个变异位点信息。
注:PI为两个随机个体具有相同基因型的平均概率,PIsibs为从一个种群中随机发现具有相同基因型的两个个体的概率。
图4 核心引物的位点频率
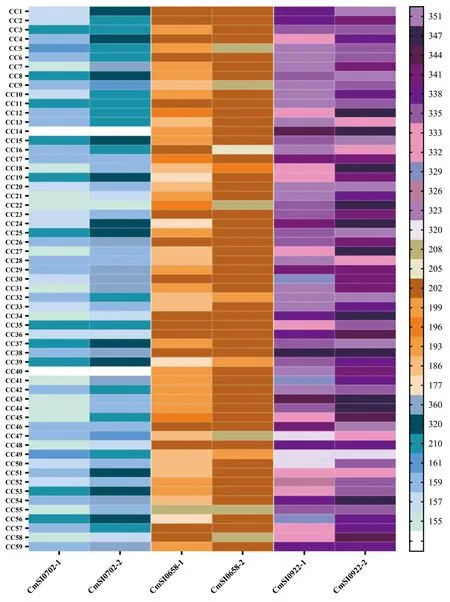
注:每种颜色框代表一个等位位点,其中白色框代表缺失位点;纵坐标为品种编号,横坐标为核心标记号。
3 讨论
3.1 日本栗品种的遗传多样性与群体结构
SSR分子标记作为第二代分子标记技术,随着基因组学和转录组学的普及,其开发利用也得到前所未有的发展,现阶段已经被广泛应用于动植物资源鉴定、物种遗传多样性以及遗传图谱构建等各种遗传相关领域[15,17]。国际植物新品种保护联盟(International union for the protection of new varieties of plants, UPOV)建议使用SSR分子标记作为构建植物DNA指纹图谱的首选方法[21]。SSR分子标记的多态性、稳定性和可重复性的好坏程度决定着相关遗传研究的有效性和成功与否[22-23]。本研究为了获得更有效的基因分型,使用了前期筛选的17对高效SSR引物[17]。17对SSR引物在59份日本栗资源中共检测到131个等位位点,每个标记平均有7.706个等位位点,位点数量高于Muller等[24]、Jiang等[25]和Terakami等[16]关于栗属植物SSR位点的报道;各基因座的PIC变幅为0.375~0.815,平均值为0.605。所有标记中有4个标记的PIC在0.25~0.50之间,表现出中等多态性;13个标记的PIC大于0.50,说明选用的SSR引物表现出高多态性,能够准确地评估供试日本栗品种的遗传特点。59份日本栗品种资源的杂合度变幅为0.389~0.857,平均值为0.685,与Nishio等[26]关于日本栗杂合度的计算结果一致,表现出较高的杂合度。这一现象是栗属植物的典型特征,究其原因是由于栗属植物为自交不亲和植物,同域内的栗属植物仅有通过杂交才能繁衍所致[17,27]。同时,日本栗杂合度略高于板栗品种的0.652[17],说明日本栗品种资源间存在较高频率的杂交事件,这一特点在日本栗群体结构的分析中也得到了验证:各个资源可以观察到有来自不同分群不同程度的遗传成分。与中国板栗品种的PIC值和香农指数(I)[17]相比可知,日本栗品种的多样性略低于中国板栗,推测有两方面的原因:其一为本试验的供试品种资源数量相比板栗品种较少;其二为日本栗进化地位较低,物种本身的多样性较低。
3.2 日本栗的分类地位与日本栗品种的聚类特征
栗属植物系统发育树的结果显示,日本栗和茅栗具有较近的种间亲缘关系,这一结果与聂兴华等[28]的研究结果一致;而Lang等[6]和Kang等[2]利用叶绿体序列聚类分析的结果则显示日本栗位于栗属植物的基部位置,出现了明显的核质冲突现象。核质冲突现象通常认为是由趋同进化、谱系分选不完全和杂交或基因渐渗三方面因素造成的[29]。栗属植物属内共7个种,其谱系分选明确,本研究推测日本栗在漫长的历史演化过程中可能保留着栗属植物祖先种的叶绿体信息,由于日本栗与其他栗属植物在历史上发生过多世代的基因渐渗,进而导致现今的日本栗核信息与日本栗祖先种已经发生了显著分化。
日本栗品种的种内系统发育树、群体结构和主坐标分析一致支持中国境内的部分日本栗品种独立形成一个分支。日本栗品种间存在着较为丰富的基因交流,尤其是朝鲜半岛的品种资源大多为混合类型,这与其所在的地理位置密切相关。
3.3 引物的鉴别能力与日本栗栽培品种(系)的指纹图谱
传统园艺果树和经济林木鉴别主要以形态分类法为主,而种内家系、杂交系、无性系等形态上较难区别,利用分子标记构建的指纹图谱能够检测到的在分子水平上的位点变异,实现简单、快速和有效的资源鉴别[30]。本研究在基因分型后,对PI和PIsibs这两个指纹识别的重要指标进行计算,使用17个SSR分子标记进行多位点匹配计算后,其值分别为3.471×10-16和7.764×10-7。Waits等[31]认为当PI约为1×10-4~1×10-2时,便可以鉴定出随机出现的大多数自然个体,表明本研究选用的SSR分子标记识别具有巨大潜力。为了快速鉴定日本栗资源,本研究根据多位点匹配分析的结果,确定了至少需要3个核心引物才能完全匹配到有唯一对应的指纹信息,并利用PIC和PI两个参数对17个SSR标记进行评估筛选,最终确定CmSI0922、CmSI00702和CmSI0658作为核心引物,3个核心引物的PI为2.256×10-4,也在Waits等[31]建议的范围之内。随后本研究利用3个核心引物构建了日本栗品种(系)资源的图像化指纹图谱,该图谱能够完全区分59份日本栗品种。现阶段中国板栗品种的指纹图谱已经构建完成[14],日本栗品种指纹图谱的补充为栗属植物的资源鉴定和保护利用提供了有力支撑。
4 结论
日本栗与中国的茅栗具有较近的种间亲缘关系,相比其他栗属植物,日本栗栽培资源分布范围较狭窄,但是其具有较为丰富的遗传多样性。此外,通过遗传分析发现中国境内的部分日本栗品种(系)相对独立,且各品种(系)间存在较为广泛的基因交流。本研究利用3个核心引物构建完成了能够完全区分59份日本栗品种(系)的指纹图谱,为今后日本栗资源的保护与利用提供了理论参考。
猜你喜欢
中国农业科学(2022年16期)2022-09-19
广西植物(2022年8期)2022-09-07
军事文摘(2022年16期)2022-08-24
中国农业科学(2022年15期)2022-08-09
舰船科学技术(2022年11期)2022-07-15
电脑报(2020年40期)2020-11-06
中学生物学(2019年7期)2019-10-17
电脑知识与技术(2018年19期)2018-11-01
新城乡(2018年6期)2018-07-09
中学生物学(2017年7期)2017-08-23
