
[噻唑基-2-14C]-毒氟磷在水-沉积物系统中的降解与残留转化研究
2022-10-29 01:33张素芬张玮玮邵思遥余志扬叶庆富
核农学报 2022年11期
吴 涛 张素芬 张玮玮 邵思遥 余志扬 叶庆富,*
(浙江大学原子核农业科学研究所/农业农村部和浙江省核农学重点实验室,浙江 杭州 310058)
毒氟磷(Dufulin),化学名N-[2-(4-甲基苯并噻唑基)]-2-氨基-2-氟代苯基-O,O-二乙基膦酸酯,是我国自主创制的新型植物抗病毒剂[1-2],属于新型的氨基膦酸酯类化合物,因其高效、低毒、低残留,且对烟草、黄瓜、番茄病毒病有良好的防治效果,而具有良好的应用与市场前景,目前已在广西、贵州等地得到推广应用[3]。研究表明,30%毒氟磷可湿性粉剂对鱼类、蜂类、鸟类、家蚕均表现为低毒、低风险特点[4]。目前,对毒氟磷的研究主要集中于作用机理、结构优化、动植物残留及其在环境中的母体降解等方面。如Shi等[5]、Zhang等[6]和Zheng等[7]研究了毒氟磷在植物(玉米、西瓜)中的降解动态与残留;Chen等[8]从手性对映体层面研究了毒氟磷在大鼠中的代谢规律;有关毒氟磷在环境中的降解行为则主要集中在母体层面[9-11]。而鲜有研究从质量平衡角度探究毒氟磷及其代谢产物在环境介质中的残留赋存转化规律。毒氟磷在水-沉积物系统中的降解动态以及残留转化规律是科学认识和评价其环境安全性的基础,然而该方面的研究迄今未见报道。
农药在环境中的行为除受自身化学性质影响外,还受到环境介质中复杂的物理、化学和生物过程(包括吸附和解吸、挥发、化学和生物降解、植物的吸收、表面径流和浸出)的影响[12-14]。水-沉积物系统是众多污染物在环境中迁移和转化的载体、归宿和蓄积库,农药在水-沉积物系统中的好氧代谢是评价其环境安全性的重要内容。因此,本试验采用14C同位素标记示踪技术,研究毒氟磷在两种水-沉积物系统中的降解动态和残留转化规律,旨在探明毒氟磷在水-沉积物体系中的消散和降解特征,同时探讨毒氟磷及其代谢物结合残留的形成规律,以期为客观评价该农药环境安全性提供基础数据和科学依据。
1 材料与方法
1.1 试验材料及仪器
1.1.1 标记化合物 [噻唑基-2-14C]-毒氟磷,由浙江大学原子核农业科学研究所和上海启甄环境科技有限公司共同合成,放射性比活度为50.06 μCi·mg-1,放射化学纯度>97%,化学纯度>99%,结构式如图1所示。
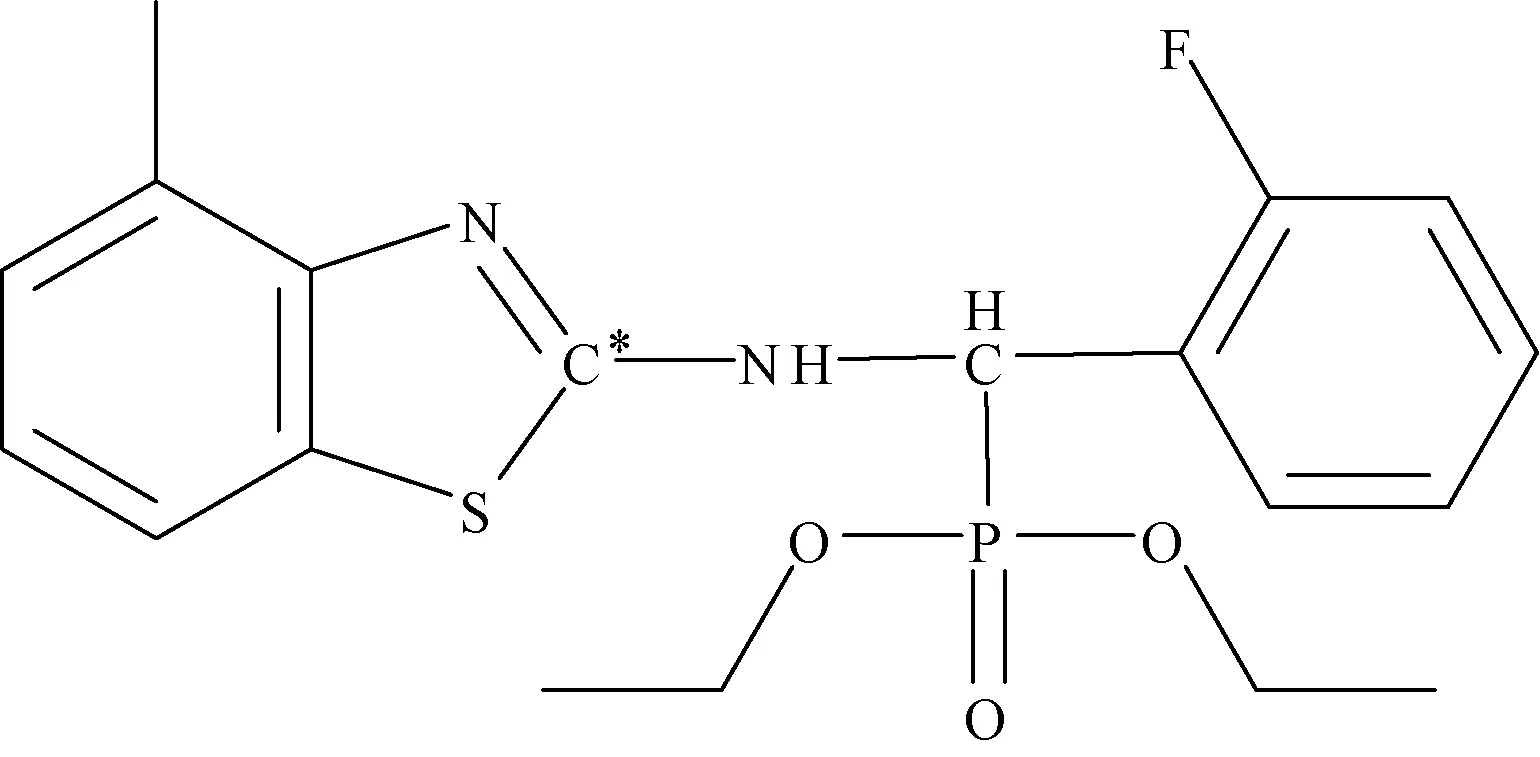
注:“*”表示14C标记位置。
1.1.2 供试水-沉积物 参考经济合作与发展组织(Organization for Economic Co-operation and Development,OECD)化学品测试准则,在有机碳含量和土壤结构不同的两块沉积物区中选择两种沉积物(近4年未被待测供试物污染),其中沉积物A取自浙江杭州启真湖(120.19°E,30.26°N),沉积物B取自浙江临安青山湖(119.82°E,30.26°N)。将沉积物静置分层后去水,再经2 mm孔径筛网过滤,备用。水和沉积物的基本理化性质见表1。
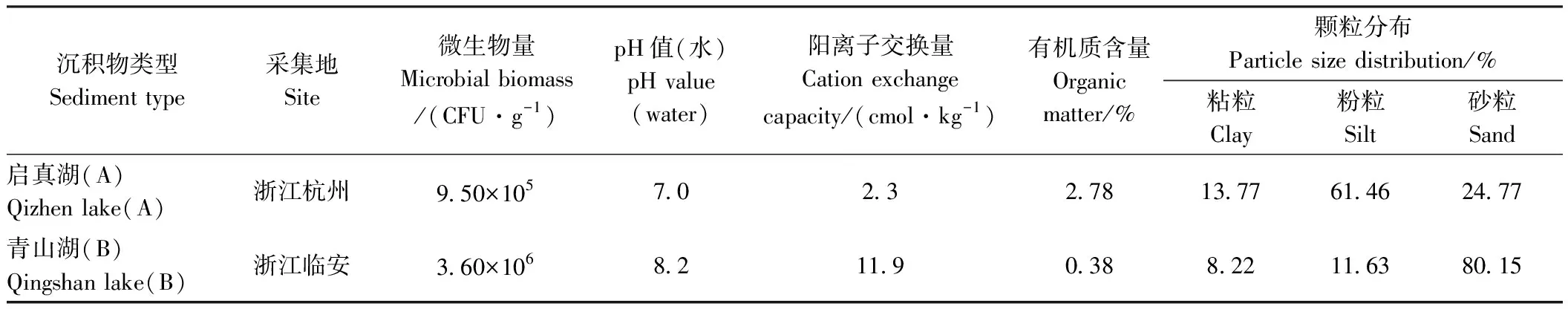
表1 沉积物理化性质
1.1.3 试验试剂 分析纯试剂:甲醇、乙腈、二氯甲烷、乙二醇、乙二醇乙醚等,色谱纯试剂:甲醇、乙腈、甲酸,均购自北京国药集团化学试剂有限公司;闪烁纯试剂:2,5-二苯基噁唑、1,4-双[2-(5-苯基)噁唑基]苯,购自上海梯希爱公司。闪烁液A、B自行配制。
1.1.4 试验仪器 Waters 2695高效液相色谱仪(美国Waters公司);PQX-450B-30H人工气候箱(宁波莱福特科技有限公司);HTC501生物氧化燃烧仪(太仓华利达公司);Tri-Carb 4910TR液体闪烁测量仪(美国PerkinElmer公司);DHG-9070A电热恒温鼓风干燥箱(上海精宏实验设备有限公司);5804/5804R高速离心机(德国Eppendorf公司);BSA224S电子天平(德国Sartorius公司);Lab-1C-80E冷冻干燥仪(北京博医康实验仪器有限公司);JPB-607A溶解氧测定仪(上海雷磁公司)等。
1.2 试验设计
1.2.1 试验装置 试验装置如图2所示,其中水-沉积物培养瓶前端的5.0 mol·L-1NaOH溶液用于吸收空气中的二氧化碳,纯水用于补充空气中的水分,培养瓶后端乙二醇/H2SO4用于吸收代谢产生的有机挥发物及碱性挥发物,0.2 mol·L-1NaOH溶液用于吸收受试物代谢产生的酸性挥发物和CO2,吸收液体积均为20 mL。
1.2.2 供试水-沉积物预培养 称取干重50 g的沉积物至培养瓶中,加入纯水180 mL,采用图2所示的装置,于20±1℃避光条件下进行预培养,培养过程中每隔1 h通空气30 min,预培养时间7 d。试验装置分为A、B两组,每组8个取样点,每个取样点设置3个重复,共48套装置。

图2 水-沉积物系统装置示意图
1.2.3 标准溶液配置 称取14C-毒氟磷10 mg,甲醇溶解定容至10 mL配置成1 g·L-1的母液Ⅰ。精确移取该母液4.59 mL,用甲醇定容至50 mL配置成91.8 mg·L-1的14C-毒氟磷标准溶液Ⅱ,14C-毒氟磷活度浓度为4.595 μCi·mL-1。
1.2.4 施药与培养 向每个培养瓶中缓慢滴加1 mL14C-毒氟磷标准溶液Ⅱ,轻微搅拌上层水相,保持沉积物静置。将培养瓶与吸收装置连接并保证气密性良好,置于20±1℃培养箱中避光培养,培养过程中每隔1 h定时通空气,每次通气时间30 min,并分别在培养后0、5、10、15、30、60、80、100 d时取样。取样前通空气10 min,通气后将培养瓶取出,分离上层水相与底层沉积物。将沉积物冷冻干燥后置于-80℃冰箱中储存。
1.2.5 沉积物可提取残留的测定 从50 g干重中均匀称取10 g干燥沉积物样品置于100 mL离心管中,依次采用体积为50 mL的0.01 mol·L-1CaCl2溶液、乙腈水溶液(V乙腈∶V水=9∶1)、甲醇、二氯甲烷进行提取[13],每种溶剂重复提取2次。具体提取方法如下:将沉积物与溶剂混匀,置于摇床上震荡提取1 h,1 000 r·min-1离心5 min,收集上清液。其中前3种提取液分别合并后定容至100 mL用于可提取残留定量分析,二氯甲烷提取液采用氮吹法吹干,再用甲醇溶解定容至50 mL。上述提取液分别取1 mL至闪烁瓶中,加入10 mL闪烁液A,避光24 h后,用液体闪烁测量仪(liquid scintillation counting, LSC)测定14C-活度,测量时间2 min,经猝灭校正曲线校正后得到测量效率(85%~90%)和放射性活度(本底40 dpm,低于2倍本底不计入数据)。采用公式(1)计算可提态残留量占引入量的比例(extractable residue,ER):
ER=(A氯化钙+A乙腈+A甲醇+
A二氯甲烷)/A引入量×100%
(1)
式中,A氯化钙为氯化钙溶液提取的扣去本底总放射性活度;A乙腈为乙腈提取的扣除本底总放射性活度;A甲醇为甲醇提取的扣除本底的总放射活度;A二氯甲烷为二氯甲烷提取的扣除本底的总放射性活度;A引入量为系统中引入的14C总活度。下同。
1.2.6 水-沉积物矿化量的测定 每5 d取下14C-CO2吸收液,并将装有新吸收液的吸收瓶连接到装置中,将吸收液稀释至80 g,混匀后定量称取2 g加入到闪烁瓶中,加入15 mL闪烁液A,避光24 h消除化学发光后用LSC测量其放射性活度的净计速率,计为Ai。根据公式(2)计算各取样点的矿化量占比(MA):
MA=(A1+A2+A3+…+An)/A引入量×100%
(2)
式中,n为吸收瓶取样的次数,Ai(i=1,2,…,n)为扣除本底的活度。
1.2.7 结合残留的测定 将经4步提取后的沉积物样品置于坩埚,研磨均匀后称取0.5 g,采用生物氧化燃烧仪将其中的放射性物质催化转化为可被闪烁液B吸收的14C-CO2,燃烧时间4 min,闪烁液B体积为15 mL,并用LSC法测量其净放射性活度。通过公式(3)计算可得沉积物样品中结合残留量占引入量的比例(bound residue, BR):
BR=ABR/A引入量×100%
(3)
式中,ABR为扣除本底的14C-结合残留活度。
1.2.8 母体降解动态研究 水相母体残留测定:取1/3水样调节pH值至3.0±0.2,用30 mL二氯甲烷萃取至上层水相无放射性检出。收集二氯甲烷相,于旋转蒸发仪上浓缩至近干,用甲醇溶解后过0.22 μm有机滤膜,氮吹浓缩定容至1 mL。采用高效液相色谱仪(high performance liquid chromatography, HPLC)对其中的放射性物质进行分离,收集母体标准品保留时间下的馏分,利用LSC测定其放射性活度,确定毒氟磷母体在水相中的含量。
沉积物相母体残留测定:对4步提取法获得的沉积物相提取液进行预处理,CaCl2水溶液调pH值至3.0±0.2,用30 mL二氯甲烷连续萃取,至上层水样无放射性物质检出,而后将二氯甲烷相转移至圆底烧瓶中旋转蒸发至近干;乙腈水溶液、甲醇与二氯甲烷提取液分别合并至上述圆底烧瓶中,旋转蒸发至近干;采用甲醇溶解瓶内放射性物质并过0.22 μm有机滤膜,氮吹法浓缩定容至1 mL。采用HPLC对其中的放射性物质进行分离,收集母体标准品保留时间下的馏分,利用LSC测定其放射性活度,确定毒氟磷母体在沉积物相中的含量。
1.2.9 HPLC分析条件 HPLC分析采用梯度洗脱的方式,洗脱程序见表2,色谱柱为C18柱(Agilent,5 μm,4.6 mm ×250 mm),柱温25℃,流速0.8 mL·min-1,光电二极管阵列(photo-diode array, PDA)检测器检测波长为270 nm。
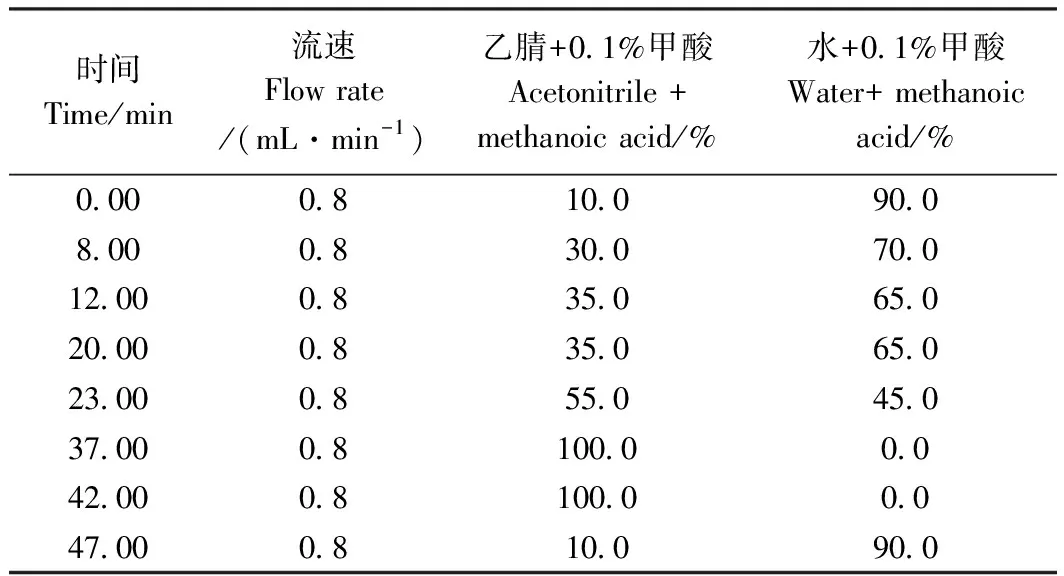
表2 HPLC梯度洗脱程序表
1.2.10 数据处理 采用单因素分析Tukey HSD对不同处理组在同一采样时间点的数据进行差异分析。所有数据以平均值±标准方差(M±SD)表示。采用Sigma Plot 14.0软件进行数据分析和作图。
2 结果与分析
2.1 14C-毒氟磷在水-沉积物系统好氧代谢中的质量平衡
本研究采用14C质量平衡法验证14C-毒氟磷水-沉积物体系中好氧代谢试验结果的可靠性。根据OECD化学品测试准则要求(OECD 308)[14],标记过的化合物总放射性回收率应在90%~110%范围内。其中14C-总放射性回收率计算如下:
14C-总放射性回收率 =(14C-矿化量 +14C-水相残留 +14C-沉积物相残留)/14C-总引入量×100%。
本试验中14C-毒氟磷在A、B两种水-沉积物系统中的14C-总放射性回收率见表3,分别为94.60%~105.36%和100.12%~109.11%,均符合OECD中关于质量平衡的要求。

表3 14C-毒氟磷在水-沉积物系统中的质量平衡
2.2 14C-毒氟磷在水-沉积物系统好氧代谢中的矿化规律
矿化率指矿化量占放射性引入量的百分比。14C-毒氟磷在两种水-沉积物系统中好氧代谢的矿化规律如图3所示。本试验条件下,14C-毒氟磷在两种沉积物系统中的矿化率均低于5%。培养初期,即培养时间<15 d,14C-毒氟磷在两种水-沉积物系统中的矿化率均较低;随着培养时间的延长,矿化率不断增加。同时,在60~100 d的培养过程中14C-毒氟磷在A系统中的矿化率显著低于B系统(P<0.05)。至培养结束时(100 d),14C-毒氟磷在两种水-沉积物系统中的矿化率分别为1.11%(A)和2.83%(B),差异性产生的原因可能与两种水-沉积物的理化性质有关。
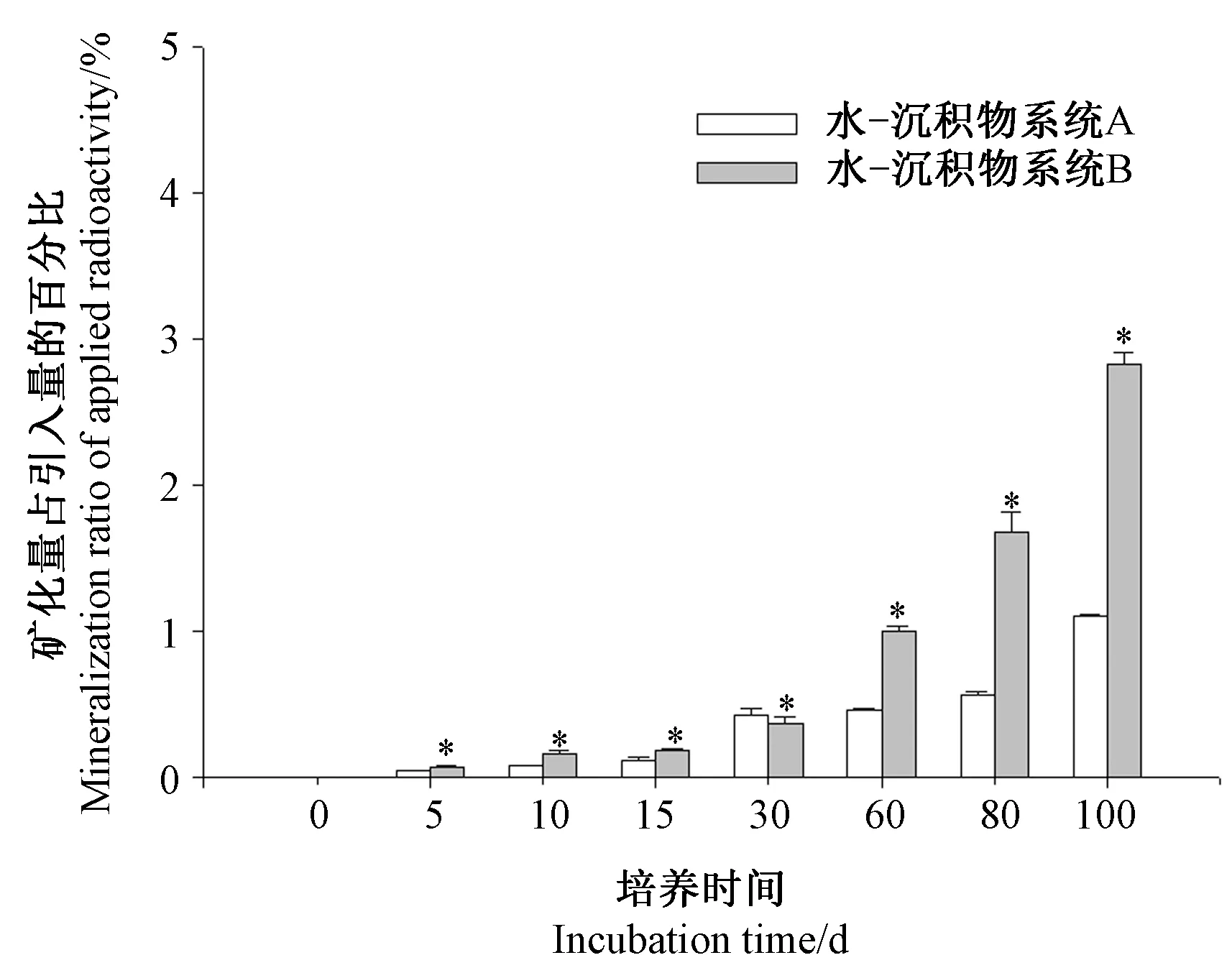
注:*表示同一时间点不同系统间差异显著(P<0.05)。下同。
2.3 14C-毒氟磷在水-沉积物系统好氧代谢中沉积物相残留的动态规律
14C-毒氟磷在水-沉积物系统好氧代谢中的可提态残留量与结合态残留量的动态规律如图4所示。由图4-a可知,14C-毒氟磷在两种水-沉积物系统中的可提态残留量随培养时间的延长均呈现先升高后降低的趋势。其中,A系统14C-毒氟磷的可提态残留量在培养80 d时达到峰值(86.51%),B系统14C-毒氟磷的可提态残留量在培养30 d时达到峰值(69.16%);至培养结束时(100 d),14C-毒氟磷在A、B两种水-沉积物系统中的可提取残留量分别下降至77.41%和43.71%。除培养初期(≤5 d)外,A系统中的可提取残留量均显著高于B系统。
由图4-b可知,毒氟磷在两种水沉积物系统中的结合态残留均随培养时间的延长呈现上升趋势。在培养前期(≤30 d),A系统中的结合残留量显著高于B系统(P<0.05)。培养至60 d后,B系统中的结合态残留量显著高于A系统(P<0.05),且呈现持续升高趋势。培养至100 d时,毒氟磷及其代谢产物在A、B两种水-沉积物系统中的结合态残留量分别达到24.11%和49.25%。在整个培养过程中,毒氟磷在两种水-沉积物系统中的结合态残留量均呈现显著差异(P<0.05)。

图4 14C-毒氟磷在水-沉积物系统中沉积物相残留动态变化规律
2.4 毒氟磷在水-沉积物系统中的降解动态规律
本试验选择一级动力学(single first order, SFO)模型计算毒氟磷母体在整个水-沉积物系统中的降解半减期(degradation time, DT50)值和水相消散半减期DT50值。毒氟磷在两种水-沉积物中的降解及水相消散变化动态如表4和图5所示。由表4可知,毒氟磷母体在A、B两种水-沉积物系统中的降解半减期分别为121.60和65.39 d,可见毒氟磷母体在B系统中更易于降解;在A、B两种水-沉积物系统中,毒氟磷母体的水相消散半减期分别为7.23和13.86 d。由此可知,毒氟磷母体在A系统中更趋于向沉积物相迁移,二者在沉积物相的可提态残留含量也同时体现了这种差异性。

图5 毒氟磷母体在水相中占比(a)和水-沉积物系统中占比(b)动态变化规律
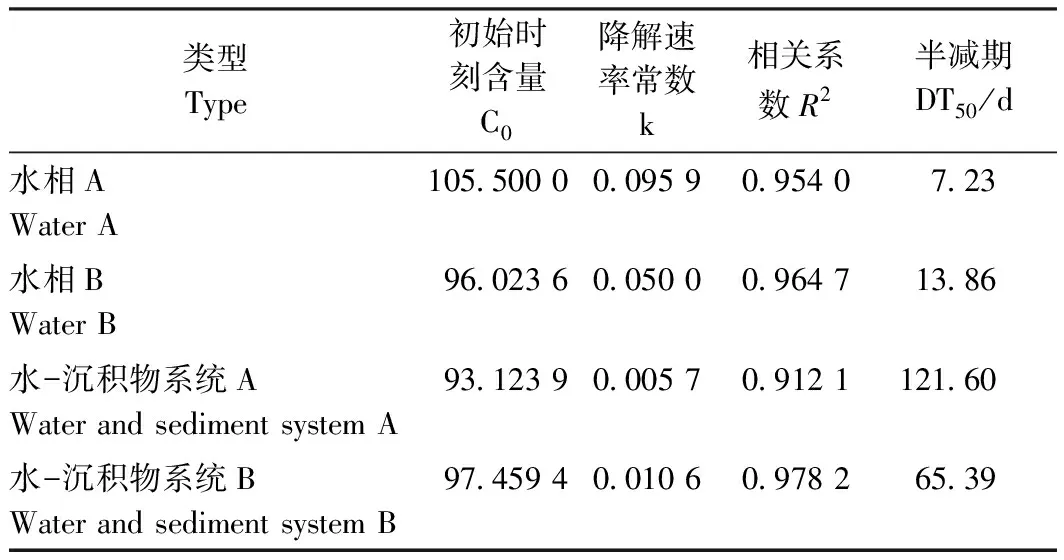
表4 好氧条件下毒氟磷在水-沉积物系统中的降解动力学规律
为了进一步跟踪毒氟磷在两种沉积物系统中不同赋存相之间的迁移规律,本试验分别对毒氟磷母体在水相和沉积物可提态残留中的含量进行了分析,结果如图5-a和图6所示。结果表明,毒氟磷母体在两种水-沉积物系统均能较快地由水相向沉积物相迁移,在本试验条件下培养至30 d时,母体在A、B两种沉积物可提态残留中的含量均达到峰值,分别占引入量的71.79%和45.44%。对毒氟磷母体在水相及沉积物相可提态残留中的含量变化进行对比,可知毒氟磷在两种水-沉积物系统中的降解主要发生在沉积物相。

图6 毒氟磷母体在两种沉积物可提态残留中的动态变化规律
3 讨论
3.1 毒氟磷在两种水-沉积物系统中的消解动态差异性
通常环境中的农药会在大气、水及土壤三相之间迁移,外界环境因素和水、土理化性质对农药的环境行为有着极其重要的影响[15]。研究农药的环境行为可为新农药的安全合理使用提供基础数据[16-17]。农药在环境中的行为受到复杂的物理、化学和生物过程(包括吸附和解吸、挥发、化学和生物降解、植物吸收、地表径流和浸出)的影响[11]。本研究中,毒氟磷在水-沉积物系统A中的水相消减速率较水-沉积物系统B更快,结合两种沉积物系统在培养初期(<30 d)沉积物相的放射性残留量(图4)以及毒氟磷降解动态(图5-b)分析可知,毒氟磷在水-沉积物系统A中更易于向沉积物相迁移,其原因主要在于A系统中沉积物对毒氟磷的吸附能力更强。土壤或沉积物等环境介质对农药的吸附能力主要与介质的理化性质有关。本研究所采用的水-沉积物系统A的沉积物有机质含量为2.78%,明显高于B系统(0.38%),由此推断本试验条件下有机质含量高的沉积物更有利于毒氟磷的吸附,这与王华子[9]关于有机质含量高的土壤更有利于毒氟磷吸附的研究结果一致。而本研究中两种沉积物的pH值和阳离子交换量差异并不是引起毒氟磷母体在水-沉积物系统A中更易于向沉积物相迁移的主导因素。
与毒氟磷母体水相消散动态不同,毒氟磷在水-沉积物系统B中较A系统更易于降解(P<0.01),降解半减期分别为65.39和121.60 d,这一现象与沉积物的理化性质密切相关。Zhang等[10]通过对比灭菌与非灭菌土壤中毒氟磷消旋体的降解半减期发现,丰富的土壤微生物含量对毒氟磷母体降解有促进作用。同时,水-沉积物系统B为弱碱性环境,而A系统pH值则呈中性,根据毒氟磷母体的分子结构,其磷酸二酯键易于在碱性环境下水解,进而影响母体在水-沉积物系统中的浓度。Zhang等[10]和Wang等[11]在关于毒氟磷的土壤降解研究中也证实了这一结论,其降解半减期与土壤pH值之间呈现良好的负相关性。因此,毒氟磷在水-沉积物系统B中的降解速率较A快,是高土壤微生物量和弱碱性环境共同作用的结果。
3.2 毒氟磷在两种水-沉积物系统中的残留赋存形态差异
通过对比毒氟磷在两种水-沉积物系统中的可提态残留、结合态残留以及矿化率的数据可知,除培养起始阶段(<10 d)外,放射性物质主要残留在沉积物相中,且矿化能力较低,其原因在于本研究所采用的示踪剂14C标记位置为噻唑基-2-14C,难以开环矿化为14C-CO2。在培养过程中,14C-毒氟磷在B系统中的矿化率显著高于A系统(P<0.05),通常农药等有机污染物在环境介质中的开环矿化主要是微生物作用的结果,在本试验条件下,毒氟磷在初始微生物含量较高的B系统中呈现了较高的矿化量,这与大多数有机污染物的矿化规律相吻合[18-22]。
结合毒氟磷在两系统中的降解规律可知,毒氟磷在A系统中降解速率较慢,半减期为121.60 d,在整个培养过程中,毒氟磷残留主要以母体化合物形式存在,而在B系统中的半减期为65.39 d,较A系统更易于形成降解产物。在培养前期(≤30 d),毒氟磷在两种水-沉积物系统中主要以母体化合物形式存在,结合态残留的形成主要与有机质含量有关,有大量研究表明,土壤有机质中含有大量能够与农药分子形成氨键、共价键、离子键等分子间相互作用的官能团,有机质含量越高,则吸附作用越强烈,更易于农药结合态残留的形成[23-27]。据此推断,有机质含量高是水-沉积物系统A在培养前期结合态残留高于B系统的主导因素。此外,作者前期通过对两种沉积物系统中的毒氟磷降解产物结构解析发现,其降解产物主要以磷酸二酯键水解产物为主,而B系统的碱性水环境则更能促进该降解过程。樊玲娥[28]关于毒氟磷水环境行为的研究发现,其在碱性水中更易降解,这与本试验结果吻合。本试验还发现,多价阳离子(如Ca2+、Al3+等)存在情况下,阳离子交换量高的沉积物B更能对毒氟磷的降解产物产生吸附作用从而形成结合态残留。通常,土壤中最活跃的组成部分为粘粒,一方面,粒径的大小决定着腐殖化的程度,粘粒粒径越小腐殖化程度越高,对有机污染物吸附能力越强,另一方面,粘粒由层状硅酸盐组成,具有较多的活性位点,能与有机污染物发生物理或化学相互作用[29-30]。Waria等[31]研究土壤中三氯生的结合态残留量时,砂土中的结合态残留量占引入量的百分比显著低于黏土,主要是由于黏土中粘粒含量较高。叶庆富等[32]研究发现14C-甲磺隆在7种土壤中形成的14C-结合态残留量在培养初期的20 d内与土壤粘粒含量呈显著正相关。刘忠珍[33]研究发现不同粒径组分对丁草胺总吸附的贡献大小不同,粘粒对丁草胺吸附贡献最大(>70%),而粉粒、砂粒的贡献相对较小。本研究所用A和B沉积物的粘粒含量相当,由此认为本研究中沉积物的粘粒含量并非是结合态残留易形成的主导因子。综上可知,毒氟磷及其降解物在水-沉积物中易与富含有机质和高阳离子交换量的沉积物形成结合态残留,且在降解速率较快的系统中更易通过降解物形式与沉积物相形成结合态残留。
4 结论
本试验采用同位素示踪技术,研究了毒氟磷在两种水-沉积物系统中的降解特征与残留转化规律。毒氟磷在两种供试水-沉积物系统中的降解特征与残留转化规律差异明显;对于整个水-沉积物系统而言,毒氟磷母体在B系统中的降解较A系统更快,而在水相中的消散规律则与之相反,碱性水环境加速母体降解;毒氟磷及其降解物在水-沉积物系统中结合态残留的形成主要受沉积物有机质和阳离子交换量的影响,其降解物更易与沉积物相形成结合态残留。
猜你喜欢
水土保持学报(2022年5期)2022-10-10
化工进展(2022年4期)2022-04-26
食品安全导刊·中旬刊(2022年3期)2022-04-15
科技资讯(2022年1期)2022-03-07
安全与环境工程(2021年5期)2021-10-08
甘肃农业科技(2020年6期)2020-07-16
分析化学(2019年3期)2019-03-30
江苏农业科学(2017年14期)2017-10-10
西部资源(2017年1期)2017-03-27
科学与财富(2017年3期)2017-03-15
