
胞红蛋白在调控细胞铁死亡中的作用
2022-10-26 03:49许明军毛健梅
中国生物化学与分子生物学报 2022年9期
许明军, 毛健梅, 习 阳
(宁波大学医学院生物化学与分子生物学研究所, 浙江宁波 315211)
胞红蛋白(cytoglobin,CYGB),又名星状细胞激活蛋白 (stellate cell activating protein, STAP),在2001年被发现于肝星状细胞中,其对于肝细胞的损伤修复具有重要的调控作用[1]。由于CYGB的结构与血红蛋白、肌红蛋白、脑红蛋白等珠蛋白相似,通过位于卟啉环中心的血红素铁对分子氧具有高亲和力,因此CYGB也被称作珠蛋白家族第四成员[2]。CYGB具有储存和运输氧气、调节活性氧/活性氮(reactive oxygen/nitrogen species, ROS/RNS)、缺氧胁迫环境下的细胞保护和肿瘤抑制等功能[3, 4]。CYGB在许多人类恶性肿瘤中普遍表达下调,过表达CYGB抑制肿瘤细胞的增殖[5]。我们之前的研究结果显示,在结直肠癌细胞中,CYGB通过p53-YAP1-ACSL4轴促进铁死亡抑制癌细胞生长[6]。
细胞铁死亡是一种新发现的调节性细胞死亡方式,被定义为一种铁依赖性的、以脂质过氧化物积累为特征的、非凋亡形式的细胞死亡[7]。铁死亡与其他调节性细胞死亡中的细胞凋亡、细胞自噬、细胞焦亡等在形态学、遗传学和代谢组学等方面存在明显区别[8]。虽然铁死亡最初在筛选癌症治疗药物中被发现,随着铁死亡逐渐进入研究者的视野,目前铁死亡已经在神经性退行性疾病、缺血再灌注损伤和心血管等疾病中被发现[9-11],探索调控铁死亡的相关机制对于疾病的预防或治疗具有潜在的应用价值。
1 细胞铁死亡
细胞内氧化还原平衡失调是细胞铁死亡的重要诱因,脂质过氧化物堆积引起细胞膜损伤是细胞铁死亡的主要特征之一。细胞内脂质活性氧及代谢物丙二醛和4-羟基壬烯醛、谷胱甘肽(glutathione,GSH)、谷胱甘肽过氧化物酶4 (glutathione peroxidase 4,GPX4)等含量及活性的改变是评价细胞铁死亡的重要指标[12]。细胞铁死亡的发生及可能的调控机制见Fig.1。
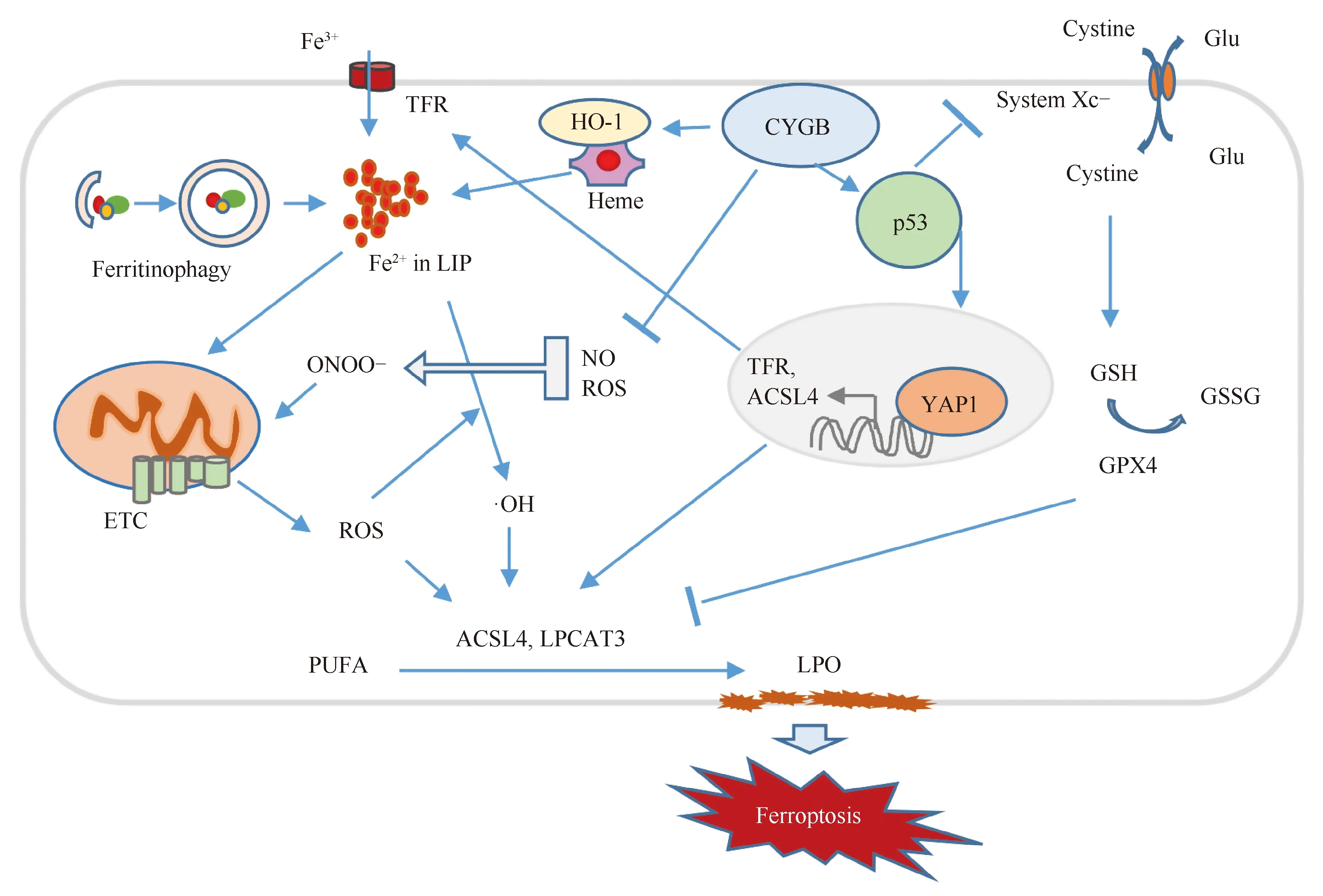
Fig.1 Diagram of regulatory mechanism of cell ferroptosis and its association with CYGB The regulation of ferroptosis by multiple pathways resulting in the oxidation of polyunsaturated fatty acid (PUFA) to produce lipid peroxide (LPO), which destroys cell membrane and induces cell death. Oxidation of PUFA mediated by acyl-CoA synthetase long-chain family member 4 (ACSL4) and lysophosphatidyl choline acyltransferase 3 (LPCAT3) is required for phospholipid peroxidation and ferroptosis. Cellular iron, which is imported by transferrin receptor (TFR), is an essential factor of ferroptosis and the autophagy-dependent degradation of ferritin (ferritinophagy). Cellular accumulated reactive oxygen species (ROS), mainly produced within the mitochondrion, react with free iron by Fenton reaction to produce hydro peroxide (·OH), which contributes to LPO production. p53 is a key regulator by activating the YAP1-mediated transcription of TFR and ACSL4 and suppressing the system Xc- for the import of cysteine, and the reduction of glutathione (GSH) results in low activity of glutathione peroxidase 4 (GPX4) to scavenge LPO. CYGB can activate p53, promote heme oxygenase 1 (HO-1) mediated heme degradation, and scavenge excessive nitric oxygen (NO) for ONOO- production
铁离子代谢及其诱导产生的活性氧在铁死亡中扮演非常重要角色[13, 14]。胞外铁离子与转铁蛋白(transferrin, TF)结合形成TF-Fe3+复合物,通过细胞膜表面转铁蛋白受体(transferrin receptor, TFR)被转移到胞内。随后,在铁还原酶前列腺跨膜上皮抗原3(six-transmembrane epithelial antigen of prostate 3,STEAP3)的作用下转变为Fe2+,由铁离子转运蛋白1(divalent metal-ion transporter 1,DMT1)将Fe2+聚集形成可变铁池(labile iron pool,LIP),多余的铁则通过细胞膜上的铁转运蛋白(ferroportin,FPN)被分泌到细胞外。细胞内的铁除了被用于氧化呼吸和酶促反应外,还能被储存在铁蛋白(ferritin,Ft)中。铁蛋白是一种具有铁氧化酶活性的铁存储蛋白质,能够将不稳定Fe2+转化为稳定Fe3+[10, 13]。一方面,增加自噬诱导铁蛋白的降解,即铁蛋白自噬(ferritinophagy)能使Fe2+上升。另一个方面,由核相关因子(nuclear factor erythroid 2 like 2,NRF2)调控的血红素加氧酶1(heme oxygenase 1,HO-1)的激活也能促使胞内血红素转化出更多的Fe2+。而高浓度游离的Fe2+与H2O2两者间发生电子转移,产生羟自由基,即芬顿反应,能使膜磷脂中的多不饱和脂肪酸(polyunsaturated fatty acids,PUFA)双烯丙基处发生链式氧化而转变为脂质过氧化物(lipid peroxide, LPO)[13, 14]。持续的脂质过氧化可直接损伤细胞膜,导致细胞功能障碍和细胞死亡。因此,由Fe2+引发的芬顿反应所产生的羟自由基,是造成细胞氧化损伤诱导铁死亡的一个非常重要原因。然而,铁除了通过芬顿反应自发诱导脂质过氧化,也可通过激活脂氧合酶的酶促反应产生脂质过氧化物[14]。细胞膜上的多不饱和脂肪酸通过氧化剂或者氧化酶类催化生成脂质过氧化物,是铁死亡的一个重要特征。首先,多不饱和脂肪酸的生物合成受长链酰基辅酶A合酶4(acyl-CoA synthetase long-chain family member 4,ACSL4)和溶血卵磷脂酰基转移酶3(lysophosphatidylcholine acyltransferase 3,LPCAT3)的调节,生成的多不饱和脂肪酸因含有双键易在氧化剂,例如过氧化氢、羟基自由基、超氧离子等活性氧存在下被氧化生成脂质过氧化物[15]。当膜上的多不饱和脂肪酸被过氧化后,其移动能力、通透性、或者与之结合的分子受到影响导致功能改变进而促进细胞铁死亡。GPX4可特异并高效地清除磷脂过氧化氢,从而抑制铁死亡。GPX4完成抗氧化作用需要消耗GSH,后者由谷氨酸、半胱氨酸及甘氨酸组成。其中, 半胱氨酸的来源胱氨酸的吸收被认为是谷胱甘肽合成的限速步骤。由跨膜转运蛋白SLC7A11(solute carrier family 7 member 11)和SLC3A2 (solute carrier family 3 member 2)组成的胱氨酸/谷氨酸逆向转运体System Xc-,可介导细胞外胱氨酸流入[16]。p53蛋白被认为是调控逆向转运体System Xc-开关和细胞铁死亡的重要分子。p53 通过直接结合SLC7A11基因启动子,并抑制其表达来减少外源胱氨酸的摄入,促进细胞铁死亡[17]。
新近研究发现,诱导细胞铁死亡是癌症治疗的一种有效方式[16]。胰腺癌细胞对吉西他滨耐药被认为是胰腺癌治疗困难的重要原因,而抑制GPX4基因表达则可逆转细胞对吉西他滨耐药,加速癌细胞死亡[18]。Erastin联合索拉非尼,可以诱导肝癌细胞内铁离子浓度增高,通过芬顿效应产生过量的活性氧,导致脂质氧化水平升高,进而诱导肝癌细胞发生铁死亡。同样,Erastin联合顺铂能提高顺铂对癌细胞的杀伤力[19]。在心血管疾病方面,Fang等[20]报道,心肌细胞中由NRF2介导的细胞铁死亡是心血管功能丧失的一个重要原因。在肝星状细胞(hepatic stellate cells,HSC)激活及纤维化中,诱导p53介导的细胞铁死亡可以改善纤维化[21]。说明铁死亡不仅在癌症的治疗,而且在纤维化,心血管疾病及代谢性疾病等的防治方面逐渐受到关注。
2 胞红蛋白
人CYGB由190个氨基酸组成,分子量为21 kD,主要以单体形式存在[1]。作为典型的珠蛋白,CYGB蛋白质核心区154个氨基酸残基(18~171位)构成非极性腔,通过G、H螺旋间狭长的缝隙与外部相连,是血红素扩散通路和配基临时结合部位,同时也是O2进出通道。CYGB由4个吡咯环,通过4个甲炔基相连形成环状,铁离子居于环中。血红素铁中的6个配位键中4个与吡咯环的N配位结合,1个与近端的F8位组氨酸残基His结合,第6个配位键被E7位的His残基占据,外源性气体型配体需要和His E7竞争,才能结合到CYGB。因此,推测CYGB除了携带和运送氧气,还可进行信号传导作用[2, 22]。
CYGB被认为是一个氧化还原状态敏感蛋白质[2]。Fe2+-CYGB与氧的亲和性较高。但在缺氧条件下,转变为Fe3+-CYGB。在CYGB氨基酸序列中具有2个半胱氨酸残基(Cys38和Cys83),能够在氧化状态下形成分子内部的二硫键(S-S)。因此,当可获取的氧的状态发生变化时,在CYGB内部的2个Cys会形成S-S,主要以Fe3+-CYGB(S-S)状态存在。此时,其对氧的亲和性下降。外源性配体结合CYGB,改变其空间结构,引起生物活性及下游信号通路的变化[23]。CYGB能结合生物膜上脂质,例如线粒体内膜上心磷脂,推测CYGB能增强脂质过氧化,影响线粒体功能[24]。事实上,当HSC激活时,细胞内CYGB表达上升,ROS含量和脂质过氧化产物增加。而CYGB被敲除后,这一现象得到显著缓解[25]。综上所述,当机体或者细胞处于氧化胁迫状态时,CYGB将参与细胞内的ROS调控及脂质活性氧代谢过程。
CYGB最初被称为星状细胞激活相关蛋白质,是HSC纤维化进程中被激活的一种蛋白质[2]。而胞内过氧化氢,脂质过氧化等活性氧物质累积是HSC纤维化的重要特征。在纤维化进程中,大量堆积的脂质过氧化代谢产物能被维生素E(vitamin E)、GSH、线粒体靶向超氧化物歧化酶(superoxide dismutase,SOD)、Mito-TEMPO等铁死亡抑制剂显著降低。我们的研究也表明,CYGB过表达后,调控肿瘤细胞对铁死亡的敏感性,说明CYGB在调控细胞铁死亡方面具有不可忽视的作用。
3 胞红蛋白与肿瘤细胞铁死亡调控
人CYGB为单拷贝基因,定位于抑癌基因富含区的染色体17q25 。近期研究表明,CYGB是抑癌基因,对维持组织器官的正常功能具重要影响[2]。小鼠cygb敲除(cygb-/-)后,在4到11个月年龄段,26%个体出现心肌肥大、肾和子宫囊肿、肝纤维化和淋巴瘤等疾病;而1年以上老龄小鼠中,约70%以上出现多个组织器官,例如肺部、肝、肠、肾及淋巴组织的自发性肿瘤[26]。在临床样本中,有研究报道,CYGB基因在乳腺癌细胞中受到甲基化影响而呈现低表达水平,恢复CYGB的表达可导致癌细胞增殖、迁移和肿瘤生长相关基因的下调[27, 28]。肺癌细胞中CYGB表达受到ΔNp63正调控,能抵抗ROS带来的凋亡,Kaplan-Meier生存曲线分析表明,ΔNp63-CYGB共同高表达的肺癌患者具有更低的生存率[29]。
Thuy le等[26]发现,cygb-/-小鼠更易被N-二乙基亚硝胺诱导发生肿瘤,并且在肝和肺组织中出现更多γH2AX阳性细胞;另一方面,cygb转基因小鼠能减少硫代乙酰胺给肝带来的损伤,说明了CYGB在抗氧化中的保护作用[2]。Fang等[30]在研究人神经胶质瘤细胞时发现,用抗霉素A作为线粒体呼吸链的抑制剂时,CYGB缺陷的细胞H2O2含量更高, 而在CYGB过表达的细胞中H2O2含量则显著降低,进一步说明,CYGB在去除ROS方面的功能。本文最近的研究发现,CYGB在肠癌细胞中具有低表达水平,而当过表达CYGB后,癌细胞生长得到抑制,线粒体膜电位受损,细胞脂质活性氧上升,且对细胞铁死亡诱导剂更加敏感。通过测序分析及对数据库中CYGB表达差异的基因富集后信号通路分析发现,CYGB能调控Hippo信号通路中的关键因子Yes1相关转录调控蛋白(Yes1 associated transcriptional regulator,YAP1),进而促进下游ACSL4表达,提升多不饱和脂肪酸含量和促进细胞铁死亡;且我们同时也证实,CYGB能提升p53表达水平[6]。而p53是调控细胞铁死亡的一个重要基因,其激活导致SLC7A11表达下调,从而抑制System XC-转运氨基酸的功能并诱导铁死亡。事实上,有报道证实,CYGB能直接结合p53,阻断其泛素化和随后的降解, 延长了p53的半衰期[31]。
诱导细胞死亡是癌症治疗的关键问题。尽管化学药物治疗取得了较大成功,但大多数癌细胞都能获得一定的耐药性,进而减弱治疗效果。在肝癌[32]、肺癌[19]、肠癌[6]和乳腺癌[33]等癌细胞中,引发铁死亡被认为是一种有效治疗方式。而对大多数癌基因或抑癌基因的调控都能影响细胞铁死亡。p53蛋白是影响细胞癌变的重要分子,在调控细胞铁死亡中具有重要作用。一方面,p53能下调SLC7A11表达,抑制逆向转运体System Xc-开关、使胞内GSH耗竭引起癌细胞铁死亡[12, 17];另一方面,p53也能激活精脒/精胺N1乙酰基转移酶1(spermidine/spermine N1-acetyltransferase 1,SAT1)的转录来促进脂质过氧化,进而促进骨肉瘤细胞铁死亡发生[34]。作为Hippo信号通路中关键因子,YAP1因被磷酸化而滞留在细胞质内,降低其细胞核活性。但在受到铁死亡诱导剂处理时,核内YAP1活性明显增强,促进靶基因TFR和ACSL4的表达,加速细胞铁死亡[35]。对影响铁死亡的重要因子的调控是增进癌症治疗效果的一种重要手段。
综合前面的研究表明,CYGB具有调控细胞内ROS水平和氧化还原平衡,以及与膜上脂质结合的特性。同时,我们也证实,CYGB对细胞铁死亡重要调控因子,例如p53、YAP1具有调控作用。鉴于小鼠CYGB敲除后多个组织器官的自发性肿瘤发生,因此,靶向CYGB同时结合细胞铁死亡诱导将会是清除肿瘤细胞的一个重要研究关注点。
4 胞红蛋白与细胞铁死亡和心血管疾病
一氧化氮(nitric oxide,NO)是调节血管张力的重要信号分子。内源性的NO由L型精氨酸在内皮细胞的一氧化氮合酶(nitric oxide synthase,NOS)催化下产生,随后进入血管平滑肌细胞(vascular smooth muscle cell,VSMC)[36]。正常生理条件下,NO结合到可溶性鸟苷酸环化酶,催化GTP产生环磷酸鸟苷,进而调节胞内Ca2+浓度和流动,引起VSMC舒张[3]。
NO失调不仅能影响平滑肌细胞和内皮细胞的增殖及死亡,也与机体的疾病状态,例如高血压、糖尿病及动脉粥样硬化紧密相关[37]。而疾病状态下,细胞会产生更多的ROS物质。NO能与ROS物质超氧阴离子(O2.-)反应,生成更具有毒性的ONOO-,引起蛋白质中酪氨酸的硝基化。一方面,ONOO-可负反馈调控四氢生物蝶呤(tetrahydrobiopterin,BH4)与内皮型NOS偶联并阻止其合成。而BH4的减少,又能促进线粒体内ROS堆积,破坏线粒体功能;另一方面,ONOO-抑制线粒体呼吸链上的复合体I/II的电子传递,以及具有清除线粒体内ROS作用的超氧化物歧化酶2功能,同时ONOO-也能促使线粒体膜孔径的变化,或者膜通透性转换孔大小的改变,使钙离子流发生改变[38]。另有研究表明,过量的NO能直接结合细胞色素氧化酶(cytochrome oxidase)而抑制线粒体呼吸链功能[3]。在心肌肥大病理模型中,线粒体促融合蛋白(mitofusin,Mfn2)表达水平下降。在缺乏Mfn2 的心肌细胞中表现出破碎的网状线粒体和改变的嵴结构,而且降低了钙依赖的线粒体通透性的改变,并且减弱了心肌细胞因缺血-再灌注损伤而引起的细胞铁死亡[39]。而大鼠和小鼠中Mfn2的减少能导致VSMC的过度增殖,加速心肌肥大[40]。说明NO代谢改变能引起线粒体变化从而诱导细胞铁死亡。在小鼠急性免疫性肝炎(acute immune hepatitis)模型中,肝细胞发生铁死亡并伴随大量RNS堆积。当用铁死亡抑制剂ferrostatin-1、iNOS抑制剂 (1400W) 或者ONOO-清除剂 (Fe-TMPyP)处理后,小鼠肝细胞中RNS和铁死亡大量减少,表明NO代谢失调与铁死亡关系密切[41, 42]。然而,值得注意的是,最近Homma等[43]在小鼠肝癌Hepa 1-6细胞中表明,NO能通过中止脂质过氧化反应来保护细胞逃离铁死亡。
CYGB具有一氧化氮双加氧酶活性,能够将NO转换成NO3-[3]。虽然肌红蛋白和血红蛋白等珠蛋白都具有清除NO的作用,但有研究表明,在VSMC中,CYGB的表达量远远超过肌红蛋白(约45倍)和血红蛋白(约200倍以上),能调控VSMC中约70% ~ 75%的NO代谢[44]。因此,CYGB通过调控NO代谢影响血管张力和血压,并通过减少ONOO-的产生保护细胞。事实上,在CYGB敲低的VSMC中及cygb-/-小鼠的血管中,NO代谢的速率显著降低,且小鼠血管壁内NO的浓度增加、血管舒张、血压和血管阻力显著降低[45, 46]。通过对动脉粥样硬化模式大鼠注射重组人源的CYGB后能显著提高SOD和GPX活性,降低血中低密度脂蛋白,提升高密度脂蛋白,改善动脉粥样硬化症状[47]。
综合前面的研究发现,cygb-/-老年小鼠出现了心肌肥大,且对NO介导的血管舒张表现增强,平均动脉血压下降了30%,说明CYGB在调控血管功能方面具有重要作用[26]。下调CYGB的表达可矫正由血管紧张素引起的高血压[3]。尽管我们的研究显示,CYGB过表达能促进线粒体活性氧物质和膜电位改变及促进细胞铁死亡。但是,在病变状态下的心肌细胞及血管内皮及平滑肌细胞处于一个什么样的氧化还原状态,同时CYGB蛋白活性状态及如何调控细胞铁死亡等这些问题仍需进一步明确。
5 胞红蛋白与肝纤维化及细胞铁死亡
肝纤维化是由HSC细胞激活,并大量增殖,表达炎症因子及细胞外基质相关基因,是导致肝硬化和肝癌的发生重要原因之一[48]。HSC激活与低氧条件紧密联系,低氧诱导因子-1α (hypoxia inducible factor-1α, HIF-1α)是低氧条件诱导下的重要因子,通过抑制线粒体三羧酸循环和氧化磷酸化,促使HSC活化。同时,细胞内ROS和脂质活性氧代谢产物增加[2]。近期的研究表明,肝纤维化与细胞铁死亡密切相关。铁死亡诱导剂Erastin及索拉菲尼能减轻小鼠肝的纤维化[49]。治疗疟疾的青蒿醚能促进p53介导细胞铁死亡,进而抑制HSC激活和肝纤维化[21]。四氯化碳是诱导小鼠肝纤维化的常用试剂,其处理小鼠同时用青蒿琥酯后,肝纤维化得到明显抑制[50]。进一步研究表明,青蒿琥酯能促进激活的HSC内铁离子富集,脂质活性氧上升,并降低抗氧化物质例如GSH含量,而这一过程又能被铁死亡抑制剂去铁胺所逆转。但是另外一个方面,高铁食物喂养小鼠能够在促进细胞铁死亡的同时提升肝纤维化,但能被铁死亡抑制剂ferrostatin-1逆转。这一结果说明,铁死亡能促进肝纤维化进展[51]。事实上在纤维化进程中,激活的HSC出现脂质过氧化代谢产物大量堆积,它们能被铁死亡抑制剂,例如维生素E(vitamin E)、GSH、Mito-TEMPO等在一定程度上减缓。说明调控细胞铁死亡在纤维化进程中具有重要影响。
虽然在HSC纤维化进程中暂无报道表明CYGB对细胞铁死亡的调控,但CYGB表达水平的变化严重影响HSC纤维化。CYGB是HSC激活后被大量表达的一种蛋白质。cygb-/-小鼠用二乙基亚硝胺处理后,肝更早出现纤维化,炎症增加,DNA断裂,大量ONOO-堆积[52]。另一方面,在cygb过表达小鼠用硫代乙酰胺处理后,肝出现中性粒细胞聚集,炎症因子大量表达和纤维化[53]。同样,cygb-/-的HSC则出现激活型HSC的特征,具有显著上升的ROS,炎症因子的表达[25]。更有报道显示,在丙型肝炎病毒感染引起人肝纤维化的不同阶段,随着纤维化进程的加剧,CYGB阳性的细胞逐渐的减少[54]。而对CYGB末端组氨酸His81突变后, 对胶原合成及自由基诱导的肝星状细胞的活化的抑制效果减弱,说明CYGB可以作为调控纤维化的一个重要靶点[55]。
不可忽视的是在HSC激活的过程中,CYGB表达被激活,而CYGB基因上游存在HIF-1α结合位点能受到低氧胁迫诱导而表达,那么CYGB是否介导HIF-1α对线粒体的调控,是否参与细胞铁死亡仍需进一步明确。另一方面,本课题组的研究表明,CYGB在癌细胞中能调控p53介导的细胞铁死亡,那么在HSC激活过程中,阐明CYGB对细胞铁死亡的调控机制将值得关注。
6 问题与展望
自CYGB发现迄今20年,对其功能的探索一直在持续。首先,作为珠蛋白家族中的一员,CYGB不仅具有携带和运输氧气的能力,还具有清除活性氧物质和调控NO代谢的作用。CYGB氨基酸序列内含有2个半胱氨酸,受细胞内氧化还原状态影响,可在分子内或者2个分子之间形成S-S键,改变CYGB与O2或者其他信号分子的结合,从而引起下游通路改变。但目前与CYGB结合的具体信号分子仍不十分明确,尤其是在一些低氧环境及病理状态下的下游信号通路仍需进一步明确;其次,尽管我们的研究表明,CYGB过表达后线粒体功能受损,且CYGB具有与线粒体膜结合的特性,但这种结合是否具有生物学意义,是否能影响线粒体对铁死亡的调控,目前仍不得而知。鉴于线粒体在调控铁死亡方面的重要作用,探寻CYGB通过何种具体方式影响线粒体功能,将会极大推动对CYGB功能的了解;最后,前面研究表明,HSC与中性粒细胞共培养条件下可激活其产生更多的ROS,且在cygb敲除后的HSC中发现了更高表达的炎症因子和趋化因子。由于炎症与纤维化、血管病变及癌症发生密切相关,那么CYGB如何影响免疫细胞,通过何种信号通路及作用机制调控炎症反应,铁死亡的调控平衡是否会是一个重要的节点,对这些问题的阐明将非常值得期待。
综上所述,本文阐明了近年来CYGB在调控纤维化、心血管疾病及癌症方面的研究进展,且将其与细胞铁死亡关联起来,并推测CYGB在细胞铁死亡调控方面具有不可忽视的重要功能(Fig.1)。因此,阐明CYGB在调控细胞铁死亡方面的机制和作用方式,将有助于进一步明确CYGB的功能,并对日益增长和加速的心血管疾病及癌症发生提供有价值的基础研究。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
海洋通报(2021年1期)2021-07-23
生物学通报(2021年4期)2021-03-16
智慧健康(2020年9期)2020-12-03
中成药(2018年9期)2018-10-09
中成药(2018年1期)2018-02-02
中成药(2017年4期)2017-05-17
现代检验医学杂志(2016年1期)2016-11-12
医学研究杂志(2015年4期)2015-06-10
中国药业(2014年21期)2014-05-26
- 中国生物化学与分子生物学报的其它文章
- PM2.5通过激活NLRP3/Caspse-1通路诱导大鼠子宫炎症反应
- “金课”背景下生物化学课程教学的创新与实践
- EHD2 Affects the Proliferation of Esophageal Squamous Cell Carcinoma by Regulating the Cyclin D1-CDK4-pRb Signaling Axis
- Therapeutic Effect of Mesenchymal Stem Cells Overexpressing Interleukin-10 on Inflammatory Bowel Disease
- miR-31改善2型糖尿病小鼠的肝损伤
- 干扰NSUN2通过调控细胞周期蛋白表达抑制黑素瘤细胞增殖