
Okur-Chung神经发育综合征1例并文献复习
2022-10-19 00:32洪锐梁张占会林舜娜李冰肖
河南医学研究 2022年19期
洪锐梁,张占会,林舜娜,李冰肖
(1.暨南大学附属第一医院 儿科,广东 广州 510630;2.暨南大学 临床医学研究院,广东 广州 510630;3.天河区妇幼保健院 儿科,广东 广州 510630)
Okur-Chung神经发育综合征(Okur-Chung neurodevelopmental syndrome,OCNDS)是一种以发育迟缓、智力障碍、言语及行为问题、面部畸形以及肌张力减退为特征的常染色体显性遗传病(OMIM#617062),该病最早由Okur等[1]在2016年发现并报道。OCNDS与CSNK2A1基因的错义或缺失/截断突变有关,该基因正常编码蛋白激酶CK2的α亚基广泛分布于体细胞中[2]。在胚胎发育时期,CSNK2A1基因敲除的小鼠胚胎会出现神经管发育缺陷、心血管畸形、胎儿水肿甚至死亡[3]。OCNDS是导致儿童生长发育迟缓及精神发育迟缓的重要病因之一[4]。全球范围内已报道的案例中,未观察到明显种族、地区、性别发病的倾向性。目前国内均为散在个案报道。本文分析了1例诊断为OCNDS患儿的临床表现及基因变异情况,并进行了文献复习。患儿家属签署书面知情同意书。本研究通过医院医学伦理委员会审批(2021-008)。报告如下。
1 病例资料
1.1 临床表现患儿,1岁,女,因“发现运动发育落后,肌张力低下6个月”就诊。第1胎第1产,足月顺产出生,出生史无特殊,出生体质量3.0 kg,5月龄常规儿童保健时发现头竖不稳,俯卧抬头小于45°,坐位过度前屈,下肢支撑差,诊断为“运动发育落后”,曾在医院行运动康复1周,未见明显效果,后未治疗。1岁就诊时,能翻身,仍不能独坐或爬,双手抓握功能差,不会主动抓握及传递玩具,无目光对视,难逗笑。否认家族中类似病史,否认家族遗传病史。体格检查:体质量9 kg(第25百分位数),头围43.1 cm(小于第3百分位数),身长70.6 cm(小于第3百分位数),体质量指数18.1 kg·m-2(第80百分位数),眼距宽,鼻梁塌,圆脸,前囟1.0 cm×1.0 cm,手指短阔,内收肌角180°,四肢肌张力低下。
1.2 辅助检查1岁Gesell发育评估:适应能力发育商17分,大动作发育商50分,精细动作发育商46分,语言发育商67分,社交能力发育商75分,总发育商得分50分。7月龄时颅脑MRI平扫示:髓鞘化延迟,双侧额颞部蛛网膜下腔增宽。见图1。

图1 患儿颅脑MRI示脑外间隙增宽
1.3 高通量全基因外显子测序北京智因东方转化医学研究中心行家系3人全基因组测序及全基因组拷贝数变异检测,并对突变基因行Sanger测序验证其家系遗传方式。基因测序结果发现患儿CSNK2A1基因存在1个c.524A>G(p.Asp175Gly)错义突变,Sanger测序家系验证来源发现该突变为新生变异,见图2。根据2019年美国医学遗传学与基因组学学会指南标准分析该变异:(1)新生变异(PS2);(2)位点位于突变热点区(PM1);(3)dbSNP数据库、千人基因组(1 000 g 2015aug_ALL)、GnomAD数据库均未收录患儿变异数据(PM2);(4)GnomAD数据库中mis_Z score≥3.09(PP2);(5)已有文献报道该位点为致病性突变[1,5](PP5)。综上,考虑该变异为致病性变异。该患儿确诊为OCNDS。
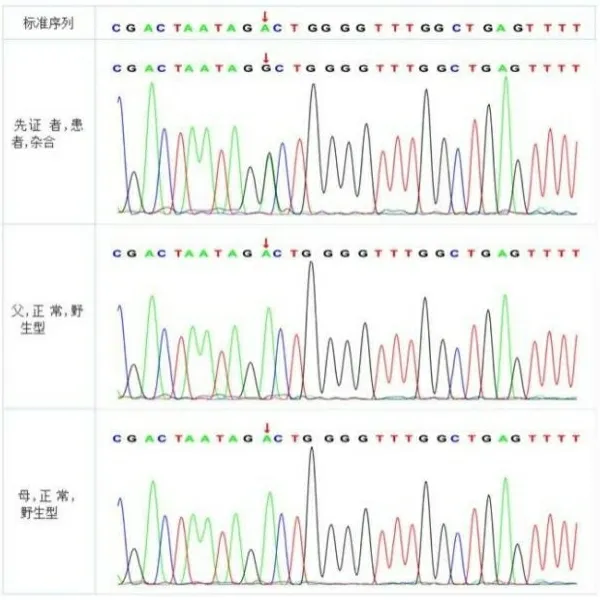
图2 家系sanger测序结果示c.524A>G为新发突变
2 讨论
蛋白激酶CK2由β和α/α’亚基组成,在体细胞中普遍表达,参与细胞增殖、存活、转录调控和胚胎发育等过程。但在不同组织上不同亚基可能具有独特的作用。CSNK2A1编码的α亚基在脑组织中高度表达,提示其与大脑功能相关,体细胞突变与多种癌症相关,而胚系变异则引起OCNDS[1]。胚系变异的CSNK2A1基因会导致CK2的α亚基活性降低及其定位紊乱,导致神经元的成熟、分化、迁移和突触可塑性降低[2],这可能是在OCNDS的颅脑MRI中常见到髓鞘发育迟缓的原因。CK2蛋白与神经系统LHX3蛋白相互作用,促进垂体发育和激素分泌,因此,垂体激素分泌异常的过程可能与CK2蛋白功能异常相关[6-7]。CK2的α亚基结构域包括ATP/GTP结合环、基本簇、活性位点及激活片段,突变常见于ATP/GTP结合环和激活片段中,此处功能域在物种间高度保守[1]。ATP/GTP结合环在维持CK2的α亚基的底物识别和ATP/GTP依赖的催化以及稳定其正常构象位移方面起着核心作用,其变异表型累及更多器官,出现更复杂神经精神障碍性疾病(neuropsychiatric disorders,NPDs)[6]。Asp175到Glu201为CK2的α亚基的激活片段,对结合磷酸化底物丝氨酸/苏氨酸残基附近的酸性残基非常重要,激活片段上已报道的突变包括p.D175G、p.R191X、p.R191Q、p.F197I、p.K198R,其中p.K198R最常见[1,8-12]。高度保守的D175WG177和G199PE201残基是激活片段形成的必需。p.D175G即甘氨酸取代天冬氨酸后,活化位点的电荷受到影响,磷酸化时干扰与Mg2+/Mn2+的结合[1]。既往认为CSNK2A1变异致CK2活性降低[13],而关于CK2的α亚基突变体CK2αLys198Arg的研究显示,主要致病机制可能不是激酶功能的丧失,而是激酶对底物的偏好出现改变,神经系统的特异性蛋白包括一些离子通道蛋白受其差异性磷酸化而失活[14-15]。针对部分受影响蛋白的研究可能为治疗带来曙光。
OCNDS罕见合并其他基因病,仅Xu等[10]报道1例中国患儿同时合并TRPS1基因变异致毛发-鼻-指(趾)综合征。CSNK2A1突变类型可为剪切变异、移码变异及错义变异,其中错义变异最常见[5,8]。尽管激酶的不同功能域发生的突变导致差异表型,但该病突变与神经发育迟缓严重程度及合并症范围的关系仍需进一步研究[2,6]。OCNDS中常见到相同变异位点而表型存在差异,如Ranganath等[16]总结的c.140G>A(p.Arg47Gln)的4例患儿出生时均有脐疝,但智力受损程度出现差别,不全合并面部畸形,2例出现行为问题,1例出现癫痫。
Okur等[1]及段浩林等[5]曾各报道1例c.524A>G(p.D175G)突变,该位点变异已证实为致病性突变[1]。患者均有显著的发育迟缓,运动发育落后,神经系统发育迟缓及面部畸形,全部合并小头畸形以及鼻、眼部问题。Okur等[1]报道案例合并听力受损及癫痫发作,本例及段浩林等[5]报道患儿均无以上发现。Colavito等[17]发现了合并视网膜病变的患者,突变位点为c.1061-1G>C ,位于CSKN2A1外显子之外,尚不明确是否由CK2α变异所致视网膜发育不良。1/3以上的OCNDS合并睡眠问题,睡眠节律改变、日夜颠倒、夜间睡眠差等问题尚未在本例中观察到。CK2参与细胞控制及信号转导过程,与睡眠节律控制相关,CK2的α亚基变异实验动物的生物钟均超过24 h,CK2的α亚基及CK2的β亚基的表达具有时空规律,尤其β亚基同样影响生物钟[2,13,18]。OCNDS患者最常见面部畸形包括小头畸形、圆脸、宽眼距、宽鼻梁、内眦赘皮、拱形眉毛、耳褶异常[1,4,8]。长期随访监测头围增长是有必要的,头围快速增加时需警惕脑积水,肌张力异常增高或出现癫痫时需尽快完善颅脑影像学检查以排查颅脑疾病。Chiu等[8]报道1例男性患儿,其生长曲线正常,6.5岁时格里菲斯心理发展量表评估结果高于平均水平,但典型的面容畸形和基因报告都证实为OCNDS,8岁时已于当地一所主流小学就读。发育延迟及精神发育迟缓是OCNDS的基本表现,年龄大于6岁时,需做智力评估同时给予适当的教育。
OCNDS临床表现主要为发育迟缓、先天畸形以及NPDs,畸形可累及全身多器官。随着年龄增长,逐渐出现NPDs或其中某些症状逐渐加重,包括癫痫发作、孤独症、注意力缺陷多动障碍、智力障碍、运动障碍、肌张力低下、睡眠问题等。部分OCNDS以NPDs首诊,超过30%的OCNDS患者存在行为问题,临床症状可疑的患儿需接受全基因外显子测序检查。对早期确诊的OCNDS需进行长期随访,尽早利用多种筛查量表评估是否出现行为问题,并转诊至儿童神经行为发育专家处诊治。目前已报道案例均为新发突变,故下一胎出现同样变异的风险很低。
猜你喜欢
分子催化(2022年1期)2022-11-02
中国农业科学(2022年16期)2022-09-19
中华实用诊断与治疗杂志(2022年1期)2022-08-31
现代仪器与医疗(2022年1期)2022-04-19
电脑报(2020年40期)2020-11-06
电脑知识与技术(2018年19期)2018-11-01
家庭百事通·健康一点通(2017年8期)2017-08-18
中外医学研究(2016年35期)2017-02-28
中国实用医药(2016年9期)2016-05-17
中学生英语高效课堂探究(2011年4期)2011-07-07
