
新生儿暴发性紫癜1例
2022-10-16 13:24李巧瑜
现代医药卫生 2022年19期
李巧瑜
(福建医科大学附属第二医院儿科,福建 泉州 362000)
暴发性紫癜(PF)是一种可致命的出血性疾病,起病前常有感染病史,对其正确认识及早期发现是降低病死率的关键因素。本院儿科收治的1例新生儿因感染导致PF的病例,现报道如下。
1 临床资料
患儿,男,出生后0.5 h。因孕35+5周早产、出生后0.5 h于2021年1月15日入住本院新生儿科。胎龄35+5周,出生史无异常,出生体重2.75 kg。其母孕期有完全性前置胎盘伴出血、妊娠期糖尿病、妊娠期高血压、妊娠合并子宫肌瘤、左侧卵巢囊肿、妊娠合并肾积水病史,否认服药史。入院查体:生命体征平稳。反应尚可,哭声尚扬。全身可见胎脂附着。头颅无畸形,前囟平软、张力不高,前囟大小1.5 cm×1.5 cm,双侧瞳孔等大等圆,对光反射灵敏。口唇红。颈软,气管居中。胸廓无畸形,未见“三凹征”,呼吸频率尚规则(52次/分),双肺呼吸音粗,双肺未闻及干湿性啰音及胸膜摩擦音。心率135次/分,律齐,心前区未闻及杂音。腹肌软,肝、脾肋下未触及,肠鸣音3次/分。四肢末梢稍冰凉,四肢肌张力正常;觅食反射、拥抱反射存在。简易胎龄评分35周。入院后给予心电监护、新生儿护理、置暖箱保暖,早产儿奶喂养,补糖,补充维生素K1预防出血等处理。患儿入院后第8天出现发热(体温37.8 ℃),心率增快,间隔4 h后开始出现皮肤瘀点、瘀斑。见图1。四肢皮肤呈大理石样花纹,查体:心率200次/分,呼吸频率63次/分,唇、舌、上腭、上牙龈黏膜,四肢、阴囊皮肤可见散在暗红色瘀点及瘀斑,以四肢为主,部分可见血疱。呼吸急促、欠规则,未见“三凹征”,双肺呼吸音粗,未闻及干湿性啰音。心音有力,律齐,各瓣膜未闻及杂音。腹软,未见肠形及蠕动波,肝肋下3 cm可触及,质中,边缘钝。四肢皮肤呈大理石花纹。四肢末梢尚温暖,毛细血管充盈时间约3 s。查血小板计数73×109L-1,C 反应蛋白 24.67 mg/L,降钙素原38.69 ng/mL,凝血全套:凝血酶原时间35 s,活化部分凝血活酶时间30 s,纤维蛋白原小于0.5 g/L,凝血酶时间42.1 s,D-二聚体大于35.2 μg/mL。
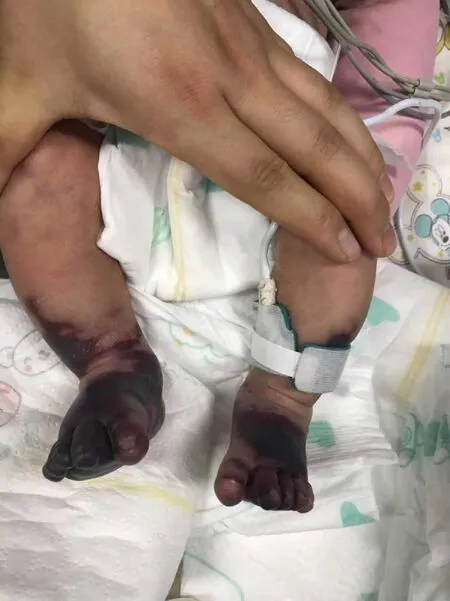
图1 入院后第8天皮肤瘀点、瘀斑
胸腹联合X线检查:(1)新生儿肺炎;(2)腹腔内肠管积气,部分肠管扩张,腹部、头颅彩色多普勒超声检查未见异常。血培养示肺炎克雷伯菌感染。脑脊液常规、生化、培养、涂片检查均未见异常。结合临床表现及实验室检查结果诊断为新生儿败血症、弥散性血管内凝血(DIC)、脓毒性休克、PF、新生儿肺炎、早产儿。给予鼻导管吸氧、美罗培南抗感染、低分子肝素钠抗凝、酚妥拉明改善循环、西地兰强心、呋塞米利尿、输注新鲜冰冻血浆、输血小板及红细胞、人免疫球蛋白加强抗感染等处理后病情逐渐好转,第6天瘀点、瘀斑较前消退,血疱基本吸收,唇黏膜、舌黏膜、会阴皮肤瘀点、瘀斑消退。第10天复查血常规、炎症指标、凝血功能和纤维蛋白原均恢复正常。患儿住院29 d后其家属要求出院,此时右下肢三趾指端仍可见暗红色瘀斑。见图2。嘱其定期至本院儿科进行皮肤护理。

图2 出院时右下肢三趾指端仍可见暗红色瘀斑
2 讨 论
PF最早发现于1884年[1],是一种罕见但极具凶险性的血栓性出血性疾病,其被认为是某些系统性疾病的标志,而不是一种疾病本身,其特征是DIC、广泛的组织血栓形成和出血性皮肤坏死。紫癜常以红斑开始,进展为不规则中心区域的蓝黑色出血性坏死,部分有血疱形成,严重者可出现肢端坏死,面临截肢,即使治愈也会留有瘢痕[2]。PF常见于儿童,发病率呈双峰型,包括1~3岁的婴儿和16~18岁的青少年,病死率较高[3]。
迄今为止,PF的发病机制尚不明确,大多数研究表明,与感染、自身免疫、药物、过敏等因素有关。根据发病机制分为新生儿PF、特发性PF和急性感染PF。新生儿PF与抗凝剂蛋白质C(PC)、蛋白质S和抗血栓Ⅲ的遗传性缺乏有关。在出生后数小时或数天内出现皮肤和其他器官大规模静脉和动脉血栓,常发生于下肢、会阴、臀部等。PC是一种维生素K-依赖性丝氨酸蛋白酶,在调节人类血栓形成方面具有至关重要的作用。PC缺乏症是血栓疾病的遗传性或后天性危险因素。遗传性PC缺乏是由位于2q14.3号染色体上的PROC基因突变所致,其突变严重的PC缺陷可出现严重的神经功能缺陷,典型表现为缺血性视神经萎缩和心室周围出血梗死,并伴有大血管血栓形成和多器官功能衰竭[4]。本例患儿在新生儿期发病,建议进行PC活性测定及基因检测,其家属拒绝,故无实验室指标证明是否存在相关蛋白缺陷。
特发性PF被认为是一种传染性自身免疫性疾病,通常在细菌或病毒感染后7~10 d内发生,与抗蛋白S抗体的产生有关,导致PS的获得性缺乏。
急性感染性PF(AIPF)是最常见的类型,往往是由产生内毒素的革兰阴性细菌感染的结果,但也可继发于革兰阳性菌和厌氧微生物或病毒感染。AIPF的主要病理生理机制是严重脓毒症导致的急性炎性反应,激活凝血系统和补体系统,导致内皮细胞功能丧失与PC的后天缺乏有关[4]。患儿可能会出现感染性休克,甚至是多器官功能障碍综合征,皮疹要么是全身性的,要么是典型的累及远端肢体并伴有快速的近端进展。AIPF主要根据典型临床表现及病原学检测诊断,临床特征:(1)全身多发皮肤出血坏死,以四肢为主;(2)发热;(3)低血压;(4)DIC。4个特征全部出现者较少见。本例患儿起病急骤,发热4 h即出现四肢皮肤瘀点、瘀斑,并迅速进展至阴囊皮肤,唇、舌、上颚黏膜等,随后出现休克、DIC、多器官功能衰竭,符合AIPF诊断标准,其后血培养提示肺炎克雷伯菌感染。
PF是一种危及生命的疾病,需要迅速识别和立即治疗根本原因和对症处理,以防止永久性残疾和死亡。在治疗方面应关注以下几个方面:(1)新生儿PF多数存在遗传性PC缺乏,以对症治疗为主,包括输注新鲜冰冻血浆、凝血因子及外源性PC替代治疗[5]。对于AIPF患儿,积极、有效的抗感染治疗至关重要,初始选择可覆盖脑膜炎耐瑟球菌、链球菌等广谱抗菌药物,合并脑膜炎者应选择透过血脑屏障良好的药物,然后按血培养药敏试验结果将针对性治疗转为目标治疗[3]。本例患儿发热早期选择了美罗培南广谱抗菌药物抗感染,待病情控制、生命体征平稳后复查炎症指标、血常规、凝血全套均正常,改用头孢哌酮钠/舒巴坦钠降阶梯治疗。抗感染总疗程2周。(2)积极对症治疗也是必不可少的,包括改善微循环、纠正水电解质紊乱、使用丙种球蛋白调节机体免疫力等。本例患儿使用酚妥拉明改善循环、西地兰强心、呋塞米利尿、人免疫球蛋白加强抗感染等。(3)若存在DIC可给予紧急输注新鲜冰冻血浆,补充消耗的凝血因子。对血小板减少症患者可输注血小板或冷沉淀治疗[6]。本例患儿发病不久即出现DIC表现,给予输注新鲜冰冻血浆、输血小板支持治疗。而肝素在PF中的治疗存在争议,但在DIC诊治相关指南中提出应进行抗凝治疗[7],早期可持续滴注肝素100~200 U/(kg·d)或低分子肝素 75 U/(kg·d)抗凝治疗。本例患儿出现DIC表现后同时给予低分子肝素钠抗凝治疗,疗程4 d。(4)当出现皮肤、肢端坏死时可进行皮肤移植术或截肢术[8]。本例患儿虽双下肢瘀斑厉害,但在精心护理及对症治疗下未出现肢体坏死。
PF是一种罕见的疾病,表现为高度血栓形成的DIC,临床判断在识别患有这种疾病的患者和制定治疗方案方面至关重要。本例患儿发现及时,处理得当,取得了较理想的效果,患儿创面愈合良好,无须给予植皮等外科治疗。
猜你喜欢
中国典型病例大全(2022年12期)2022-05-13
健康体检与管理(2021年5期)2021-09-10
时代英语·高一(2021年1期)2021-03-18
初中生学习指导·提升版(2020年5期)2020-09-10
时代英语·高一(2020年1期)2020-05-15
特别健康·下半月(2020年5期)2020-05-15
时代英语·高一(2019年1期)2019-03-13
数理化学习·高三版(2015年3期)2015-10-21
党员文摘(2009年8期)2009-08-22
职业·中旬(2009年12期)2009-06-01
