
等离激元共振光转热增强负载纳米金对丁二烯选择性加氢的催化性能
2022-10-14 09:41李学宇王朝陈雅李可可李建全金顺敬陈丽华苏宝连
高等学校化学学报 2022年10期
李学宇,王朝,陈雅,李可可,李建全,金顺敬,陈丽华,苏宝连,4
(1.武汉理工大学材料复合新技术国家重点实验室,武汉 430070;2.郑州大学河南省高温功能材料重点实验室,郑州 450052;3.襄阳职业技术学院,襄阳 441050;4.那慕尔大学无机材料化学实验室,比利时那慕尔B-5000)
烯烃,尤其是纯度大于99.999%的C2~C4烯烃,是工业催化聚合反应制备聚合物、表面活性剂等有机化学化工产品的基础化学物质[1,2].目前,这些单烯烃原料主要由原油加氢裂化后得到[3],同时会产生少量的炔烃和二烯烃杂质,如,丙烯原料中有2%~8%(质量分数)的丙炔和丙二烯杂质;丁烯原料中有0.3%~0.8%(质量分数)的丁炔和丁二烯杂质[4].这些杂质会毒害用于单烯烃聚合的催化剂,并通过将催化剂转化为稳定的中间体而终止聚合反应[5][如2-丁烯和丁二烯与Cp*2ScCH3(Cp*=η5-C5Me5)反应,生成稳定的Cp*2Sc(CH3)=C(CH3)2化合物[6]].因此,工业上对化工原料的纯度要求较高,杂质浓度必须控制在10-5以下[7].
选择性催化加氢可以选择性地将炔烃和二烯烃杂质转化为有价值的单烯烃原料[7,8],是单烯烃聚合工业中除去炔烃和二烯烃杂质最重要的途径.目前,Pd基双金属催化剂(如Pd-Ag/Al2O3[9~11])是主要的商用催化剂.但Pd基催化剂在高转化率(>80%)下,对产物单烯烃选择性(<20%[12])较低.此外,在催化加氢过程中Pd表面附近相邻的烯烃分子容易发生聚合反应,使得Pd表面易发生积碳而失活[13],很大程度上限制了其高效利用.因此,开发一种高活性和高稳定性兼备的选择性加氢的催化剂很有必要.
Au基催化剂具有高单烯烃选择性及较好的催化活性和稳定性,在选择性催化加氢反应中具有很大的应用潜力[14~16].与Pd基[12]、Ni基[17]等炔烃或二烯烃加氢催化剂相比,Au基催化剂可以在高转化率下,保持稳定的高单烯烃选择性,而完全转化时所需的反应温度相对较高.如,Zhang等[12]通过浸渍法制备的Pd/Al2O3催化剂,在52℃下的丁二烯转化率为98%,但丁烯选择性却急剧下降至零.Wang等[17]利用尿素沉积法(DPu)制备了Ni/TiO2催化剂,当反应温度升高至160℃时,对1,3-丁二烯的转化率可达95%,但此转化率下的丁烯选择性仅为46%.然而,Derrouiche等[14]以同样的DPu方法制备的Au/ZnO催化剂,对丁二烯的转化率达100%时,可以稳定地保持高丁烯选择性(100%),但完全转化率下所对应的反应温度高达180℃.同样,Masoud等[16]通过在溶液中用硼氢化钠还原制备的Au/SiO2催化剂,在96.7%的丁二烯转化率下的丁烯选择性为100%,而此转化率下所对应的反应温度(240℃)远高于Pd基和Ni基催化剂.可以看出,虽然Au基催化剂在炔烃/二烯烃完全转化下,具有很高的单烯烃选择性,但是Au基催化剂目前主要亟待解决的问题是,炔烃/二烯烃高转化下所需的反应温度过高[14,16,18~20],远高于烯烃工业中所能提供的温度(40~140℃[21]).因此,实现Au基催化剂绿色低能耗驱动选择性催化加氢的意义重大.
光热转换是一种绿色、高效且极具潜力的能源利用方式.太阳能作为地球上储量最大的清洁能源之一,将太阳能高效转换为热能的光转热技术在近年来备受关注.等离子体金属纳米颗粒是光转热应用中最重要的一类基础材料.当金属缩小至纳米尺度(Au尺寸为1~100 nm[22])时,在光的照射下纳米颗粒表面的简谐振动电子云会与光波发生协同共振,进而吸收光子能量,这部分能量以产生热电子的形式衰减,随后热电子释放出大量热能,实现光热转换,该过程为等离激元共振光转热[23,24].目前,这一原理已被应用在CO2加氢等催化反应中,Wang等[25]将Au纳米粒子负载在ZnO催化剂表面,用532 nm的激光激发后产生了明显的热效应,可使CO2和H2转化为CH4和CO,证明了等离子体纳米颗粒的光转热效应在CO2热催化应用中的可行性.Cronin等[26]研究了Au/TiO2催化剂对CO2光还原的催化性能,并通过改变光波波长来调控产物的分布.结果表明,532 nm光波激发主要产生CH4,254 nm的紫外光激发产生乙烷和甲醇等其它产物.同时,Zhang等[27]利用Au/CeO2催化剂的光热效应,实现了高转化率和选择性的光热反水煤气变换,结果表明,光热反应速率比热条件下高10倍,且保持稳定的活性和选择性.
此外,在选择性催化加氢领域也有类似应用,张铁锐等[28]通过冷冻干燥辅助法在掺氮石墨烯上负载了Pd单原子,主要利用掺氮石墨烯载体的高吸光性,从而驱动负载金属表面的乙炔选择性催化加氢反应过程.结果表明,施加0.54 W/cm2的光照时,掺氮石墨烯载体为Pd单原子加氢反应提供了125℃的反应温度,实现了乙炔选择性加氢的高催化活性,但稳定性(<24 h)较差.值得注意的是,该研究利用的是载体的高吸光、热振动实现的光转热,加热的活性中心驱动催化反应.相比较而言,直接利用催化活性金属纳米粒子作为等离激元共振光产热,实现光照自加热,从而自驱动表面催化反应可能具有更高的研究价值.在工业高能耗的不饱和烯炔烃选择性反应方面,相关研究还处于起步阶段,探究和开发以相应金属催化活性中心等离激元光转热驱动的催化反应过程,是推进当前工业催化加氢绿色低能耗的重要途径之一.
本文基于金纳米颗粒的等离激元共振光转热效应,结合具有高光吸收性质的氧化石墨烯载体,通过阳离子吸附法制备了氧化石墨烯负载纳米金催化剂(Au/GO),并以丁二烯选择性催化加氢为探针反应,探究了光照条件下金属纳米粒子的等离激元光转热效应对其选择性催化加氢性能的增强机制.
1 实验部分
1.1 试剂与仪器
无水乙醇和乙二胺(C2H8N2,纯度>99.5%),均为分析纯,国药集团化学试剂有限公司;氯金酸(HAuCl4)、氧化石墨烯(GO,纯度>99%)和Nafion全氟磺酸树酯溶液(质量分数5%~5.4%),分析纯,阿拉丁试剂上海有限公司;去离子水,武汉品管仪器设备有限公司Millipore系统.
Bruker D8 Advance型X射线衍射仪(XRD),德国布鲁克AXS公司,利用Scherrer方程[粒径Dc=0.89λ/(B·cosθ),其中,λ为X射线波长,B为衍射峰半高宽,θ为衍射角]计算平均Au纳米颗粒粒径;Hitach S-4800型场发射扫描电子显微镜(SEM),日本日立公司;JEOL JEM 2100F型透射电子显微镜(TEM,日本电子株式会社),粒径统计公式为dAu=Σnidi/Σni(式中:ni为相同粒径的颗粒数目;di为粒径,nm);Lehman-Merbu Prodigy 7型电感耦合等离子体光谱仪(ICP),美国利曼-徕伯斯公司;Varian Excalibur Series 3100型傅里叶变换红外光谱仪(FTIR),美国赛默飞世尔科技公司;Lambda 750 S型紫外-可见近红外分光光度仪(UV-Vis),美国Perkin-Elmer公司;PHI5000 Versaprobe III型X射线光电子能谱仪(XPS),日本ULVAC-PHI公司,原位测试在0.2 W/cm2的光照下进行;RENISHAW-InVia(M005-141)型拉曼光谱仪(Raman),英国雷尼绍公司.
1.2 实验过程
1.2.1 Au/GO催化剂的制备根据阳离子吸附法[29]制备Au/GO催化剂.具体步骤如下:将HAuCl4溶液(4 mL,0.4 mmol/L)和16 μL C2H8N2混合,在45℃下搅拌2 h,获得金前驱体溶液.将20 mg GO预先分散在20 mL去离子水中,然后将1 mgAu/mL不同体积(40,100,200和400 μL)的金前驱体溶液加入到GO溶液中,搅拌10 min混合均匀.随后用去离子水反复离心洗涤5次(离心转速10000 r/min,每次持续15 min).最后将收集的固体分散在10 mL去离子水中,进行冷冻干燥24 h,将干燥后的固体在180℃空气中煅烧2 h,即得到金负载量(质量分数)分别为0.2%,0.5%,1%和2%的Au/GO-x催化剂(x=0.2,0.5,1和2).由ICP测试得出,Au/GO催化剂的实际金属负载量(质量分数)依次为0.15%,0.46%,0.92%和1.88%.值得注意的是,由于溶液中金离子在光照下容易还原,实验制备过程需在暗环境下操作.
1.2.2 催化加氢性能测试 丁二烯的选择性催化加氢反应在间歇式反应器中进行.首先将10 mg催化剂以乙醇为溶剂涂敷在反应器中的玻璃片上,在60℃烘箱中烘干5 min.随后将反应混合气体(质量分数分别为0.03%丁二烯、15%丙烯、25%氢气和59.97%氦气)引入并截断到常压反应器中,用氙灯(PLS-SEX300,工作电流为10~21 A)照射,并分别改变光照强度(0.1~0.2 W/cm2)和反应时间(0~90 min)进行光热催化加氢反应.反应结束后,用氦气将反应器中的气体推入气相色谱仪[Perichrom PR 2100型离子火焰检测器(FID,武汉泰特沃斯科技有限公司)温度为220℃,色谱柱为毛细管柱,柱箱温度为75℃]进行分析.丁二烯转化率(Conversion,%)和丁烯选择性(Selectivity,%)的计算公式如下:
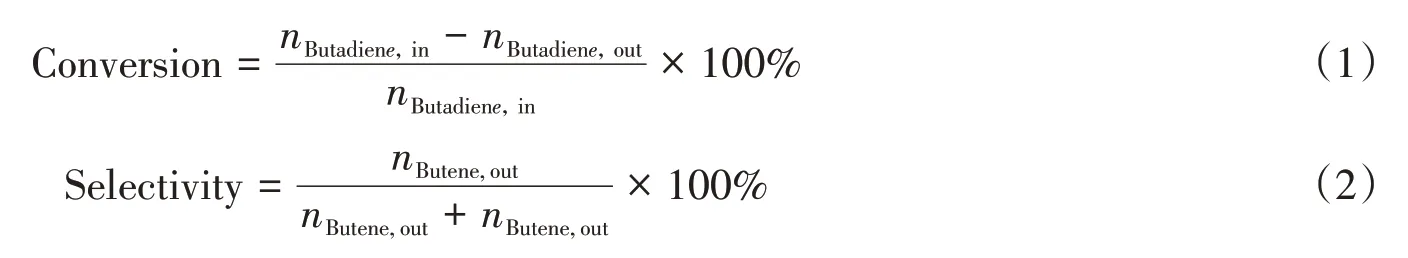
式中:nButadiene,in和nButadiene,out(mol)分别为进入和排出反应器的丁二烯摩尔数;nButene,out和nButane,out(mol)分别为排出反应器的丁烯和丁烷的摩尔数.
由于生成NH3·H2O而没有离开反应体系致使溶液呈现碱性,从而使CO2也很难从溶液中逸出,造成NH3·H2O和CO2并存于同一体系中,使水解反应不能进行彻底而很快达到平衡,反应进行的程度很小,为此两者可以大量共存,这一点与和Al3+的反应不同。
为了探究Au纳米颗粒粒径与催化加氢活性的关系,基于在0.2 W/cm2的光强条件下测得的催化加氢转化率,计算了催化加氢活性(Activity,molButadiene·s-1·g-1Au):

式中:nButadien(emol)为在0.2 W/cm2光强条件下在Au/GO催化剂催化下被转化的丁二烯摩尔数;mA(ug)为负载纳米金的实际质量;(t5400 s)为反应气体与催化剂的接触时间.
为了对比光-热驱动催化和热催化的加氢性能,将Au/GO-0.5催化剂在100℃条件下进行热催化加氢测试,反应气体与光热催化测试中相同,进行间歇式反应90 min,用气相色谱仪检测.
1.2.3 光热转换测试 为了探究Au/GO样品的光转热能力,将10 mg样品放置在反应器中,通过调节不同的光照强度(0.1~0.2 W/cm2),照射10 min后,使用红外热成像仪测量温度.
纳米金的等离激元光转热效率主要是利用短时间内的温度变化来计算.假设在环境条件相同的情况下,初始温度变化的热损耗忽略不计,催化床各处的温度相同.将7 mg Au/GO催化剂分散在2 mL去离子水中.在0.2 W/cm2的光照下,用ST9450型红外成像仪(上海雨沃仪器设备有限公司)测定催化剂的光热转化温度.光热转换效率(η,%)的计算方法如下[30]:
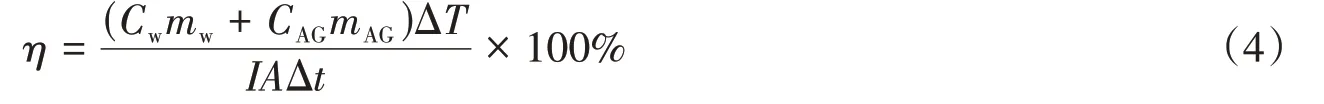
式中:C(w4.18 kJ·kg-1·K-1)和CAG(kJ·kg-1·K-1)分别是水和Au/GO催化剂的比热容;mw和mA(Gkg)分别是水和Au/GO样品的质量;ΔT(℃)是Δt时间内升高的温度;A是有效光照面积,为5.3 cm2;I是光照强度,为0.2 W/cm2.由于测试中样品的质量(0.01 g)相比水的质量(2 g)很小,即可假设CAGmAG值近似为0.
1.2.4 光生电子测试 为了揭示不同金属负载量样品间等离子体效应产生的热电子差异,在0.2 W/cm2光强条件下进行光电流测试.首先,将5 mg催化剂分散在25 μL乙醇、25 μL水和5 μL全氟磺酸树脂溶液的混合溶液中,超声处理5 min.随后,将样品的浆液涂抹在导电玻璃上并进行干燥.光生电子测试在一个标准的三电极体系中进行,使用导电玻璃(3 cm×1 cm)作为工作电极,铂电极作为对电极,Ag/AgCl标准电极作为参考电极,0.5 mol/L Na2SO4溶液作为电解质,在0.5 V的极化电位下评价样品的瞬时光电流.
2 结果与讨论
2.1 催化剂的表征
图1是GO粉末载体和Au/GO催化剂的XRD谱图.由图1(A)可见,GO在2θ=11°处有一个强衍射峰,与石墨粉在2θ=26.6°处的衍射峰相比,这是由于碳结构在氧化剥离时发生了晶格畸变,并有大量官能团并入石墨的层间,改变了石墨的固有结构[31].负载纳米金后[图1(B)],Au/GO样品在2θ=25°附近有一个明显的宽峰,表明GO的层间间距减小,GO从单层状态向多层结构变化[32];此外,2θ=38.2°处的衍射峰对应Au(111)晶面,表明Au纳米颗粒在成核生长的过程中(111)晶面相对稳定,是其主晶面.利用谢乐公式对XRD谱图中Au衍射峰进行计算,可以获得Au纳木颗粒的平均粒径.经过计算得出,随着金负载量由0.2%增加至2%,负载型纳米Au颗粒的尺寸逐渐由13.6,15.1,17.9 nm增大至20.5 nm.
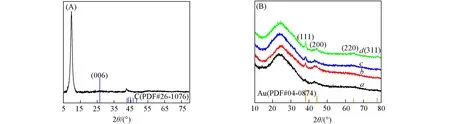
Fig.1 XRD patterns of GO(A)and Au/GO catalysts(B)

Fig.2 SEM images of GO(A),Au/GO-0.2(B),Au/GO-0.5(C),Au/GO-1(D),Au/GO-2(E)
为了进一步获得Au/GO催化剂的微观结构信息,对还原后的样品进行了高分辨透射电子显微镜(HRTEM)表征.由图3(A)可观察到,GO载体本身是典型的层状结构.纳米金修饰后[图3(B)~(E)],GO载体仍然保持层状结构.随着Au负载量的增加,Au纳米颗粒尺寸逐渐变大(10.78~21.47 nm)[图3(G)~(J)],且没有发现明显的团聚现象.统计结果表明,0.2%,0.5%,1%和2%Au负载量的纳米颗粒尺寸分别为(10.78±1.1),(15.20±0.9),(17.03±1.2)和(21.47±2.4)nm.上述结果与XRD谱图计算所得的数据基本一致.由图3(F)插图中Au/GO-0.5催化剂表面Au纳米颗粒的晶格条纹可见,晶面间距为0.235 nm,对应Au(111)晶面[33],这与XRD谱图的结构一致.由此可以得出,通过阳离子吸附法制备的Au/GO催化剂中,负载的Au纳米颗粒尺寸主要分布在10~22 nm范围内,且多呈球状.
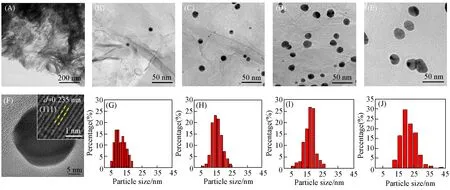
Fig.3 TEM images of GO(A)and Au/GO-0.5 catalyst(F),TEM images(B―E)and the particle size distributions(G―J)of Au/GO-0.2(B,G),Au/GO-0.5(C,H),Au/GO-1(D,I)and Au/GO-2(E,J)
图4的红外和拉曼谱图解析了负载金对载体(GO)表面官能团和缺陷的影响.图4(A)中,GO样品在3404 cm-1处有明显的红外吸收峰,这主要是因为—OH伸缩振动模式的存在,表明在GO载体中存在大量的羟基;1054 cm-1处的峰为醇或酚的C—O—C伸缩振动;1726 cm-1附近的峰对应的是C=O弯曲振动;1619 cm-1处对应的是RC—CR′的吸收峰.负载不同量的金属后[图4(B)],在3404 cm-1处的峰强度发生不同程度的降低.此外,图4(C)的拉曼光谱中GO的两个特征峰[约1353 cm-1(D峰)和约1577 cm-1(G峰)]强度比(ID/IG)可用于评价缺陷程度.可以看出,GO载体的氧化程度最高(ID/IG约0.95),随着金属负载量的增加,载体的氧化程度略有下降(Au/GO-2的ID/IG约为1.03),表明Au与部分含氧官能团发生结合;同时在2710 cm-1附近并未观察到还原氧化石墨烯的特征峰(2D),表明负载Au后的GO并未转变为还原氧化石墨烯,而是层间间距减小,单层逐渐转变为多层.从图4(A)~(C)的测试结果可以看出,GO载体表面存在大量的含氧官能团,这些含氧官能团促进了Au离子的成核和生长[34],与Au粒子的强相互作用使得负载金纳米颗粒具有很好的分散性.
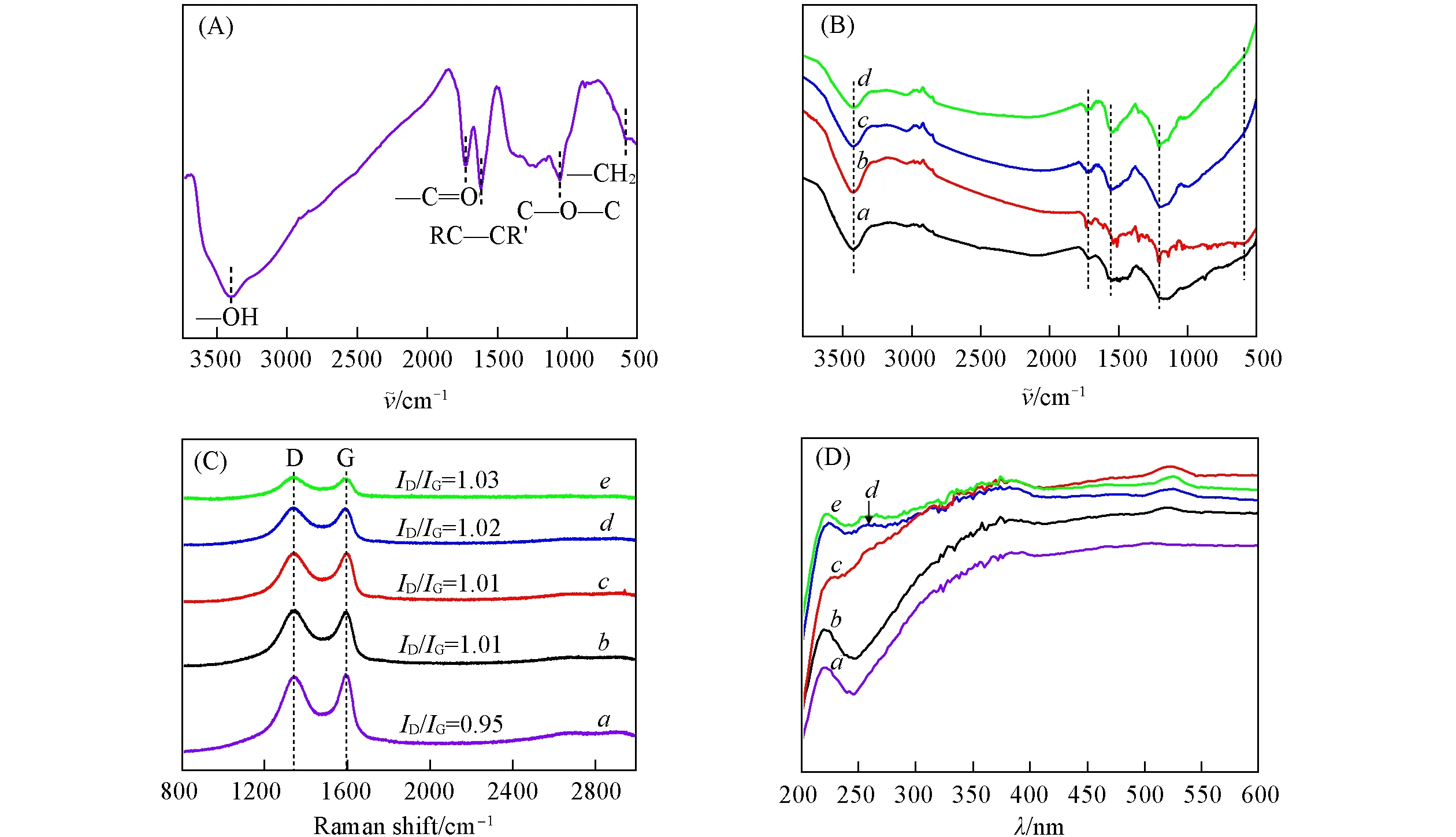
Fig.4 FTIR spectra of GO(A)and Au/GO catalysts(B),Raman(C)and UV-Vis(D)spectra of GO and Au/GO catalysts
图4(D)为GO和Au/GO样品的紫外-可见光谱.从图中可以看出,5条谱线在230 nm波长处均有一个明显的吸收峰,这表明碳六元环碳碳双键(C=C)的π-π*电子跃迁.与GO相比,负载金纳米颗粒后的样品在230 nm处的峰并未发生明显偏移,表明负载前后GO载体的氧化程度略有下降,但共轭电子结构并未发生明显变化[32].同时,负载金纳米颗粒后的样品在520 nm附近均有一个明显的特征峰,这对应着Au纳米颗粒的特征吸收峰,且峰的强度和位置随Au纳米颗粒的尺寸和负载量发生轻微的变化和红移.由图4(D)可知,与GO样品相比,Au/GO样品对300~600 nm的光存在较强的吸收,其中,Au/GO-0.5样品比其它样品对光的吸收更高.
图5是Au/GO样品的XPS谱图.可见,83.9和87.6 eV分别为Au4f7/2和Au4f5/2的特征峰,与金属Au0态的特征峰一致[35].值得注意的是,尽管增加了Au的负载量,Au4f7/2的峰位置在样品中基本没有变化,表明不同金属载量的催化剂样品中Au纳米颗粒的电子态相似,Au纳米颗粒的电子状态与负载量无关.
2.2 Au/GO样品的光热转换效率
为了研究Au/GO样品的光热转换效率,对GO和Au/GO样品进行了光热转换温度的测试.具体的光热转换温度曲线如图6(A)所示,可以看出,GO和Au/GO样品在没有施加光照和外部热源条件下的宏观温度均约为20℃,光照时样品表现出不同程度的温度变化.其中,作为对照组的GO载体在0.1~0.2 W/cm2的光照时,光热转换温度从50℃升高至63℃,这主要是因为多层GO黑色且具有高吸光率,受到光照时可以促使碳原子振动而吸收太阳能,并以热量的形式释放出来[36].负载纳米金后,Au/GO-1和Au/GO-2样品在受到0.1~0.2 W·cm2的光照时,表现出的光热转换能力最高,从80℃升高至110℃;其次是Au/GO-0.5(80~100℃)和Au/GO-0.2(80~95℃)样品.这是由于等离激元共振效应受到纳米粒径的影响,在10~40 nm范围内,粒径越大,等离子体共振越强,光热转换温度越高[37],结合TEM的粒径统计分析得出,Au/GO样品的光热转换温度符合此规律变化.因此,GO负载纳米金后的光热转换温度比GO要高得多,这表明负载在GO表面上的Au纳米颗粒受到光照后,发生了等离子体共振效应,Au纳米颗粒和GO载体的共同作用实现了光热转换.图6(C1)~(C5)是Au/GO-0.5样品在受到不同光照强度时,表现出的瞬时光热转换温度,可以判断出反应器中的温度是从催化剂向周围环境传递产生的,这表明催化反应的驱动力来自光热催化剂自身的光热转换.
为了计算样品的光热转换效率,测量光热催化剂样品在一定时间内的温升梯度.由图6(B)可以看出,对于纯水体系(2 mL),在0.2 W/cm2光照强度条件下,随着光照时间的延长,300 s后水温从室温(20℃)升高至32℃.向纯水中加入7 mg GO,在相同的光照时间下,温度由室温升高至44℃.在GO载体上修饰纳米金后,发现光转热温度大幅提高,并且随着金负载量的增多而升高,其中Au/GO-2样品的温升(34℃)变化最大.这表明光照条件下负载的纳米金表现出良好的光热转换效应,且随着不同负载量的纳米金粒径的增大,光热转换能力逐渐增强.为了更好地量化光热转换效率,选取0.2 W/cm2光照强度下,200 s内的温度变化计算了光热转换效率(η)(表1).GO单独产生的光热转换效率为50%.当GO表面负载Au纳米颗粒后,Au/GO样品的光热转换效率分别为72%,76%,80%,88%.以上结果表明,Au/GO样品受到光照时,在Au纳米颗粒的等离子体效应和GO载体的高吸光率共同作用下,实现了太阳能的高效转换.

Fig.5 XPS spectra of Au/GO-0.2(a),Au/GO-0.5(b),Au/GO-1(c)and Au/GO-2(d)
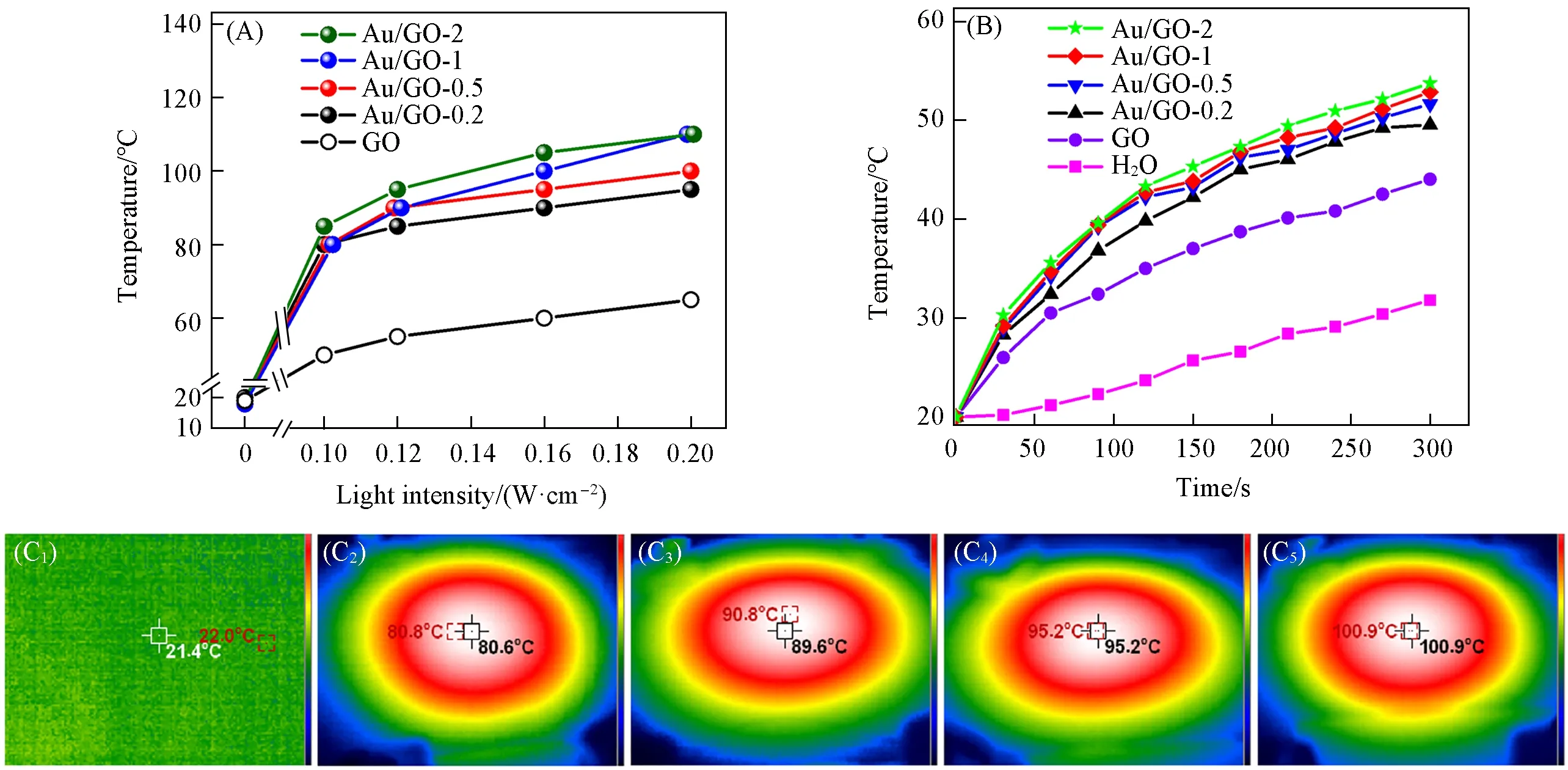
Fig.6 Surface temperature profiles of Au/GO catalysts photothermal conversion(A),and bulk transient temperature profiles of Au/GO catalysts heated under 0.2 W/cm2(B)and thermal imaging temperature of Au/GO-0.5 catalysts with different light intensities(C1―C5)

Table 1 Photothermal conversion efficiency of H2O,GO and Au/GO catalysts within 200 s at 0.2 W/cm2
2.3 Au/GO光转热驱动的丁二烯选择性催化加氢性能
图7(A)和(B)给出了在不同光照强度下Au/GO催化1,3-丁二烯的选择性加氢性能.随着光照强度增大,Au/GO催化剂的丁二烯转化率逐渐升高,且丁烯选择性均在90%以上.其中,在0.2 W/cm2光照下,Au/GO-0.5催化剂表现出最高的催化活性(丁二烯转化率为99%,光转热温度为100℃).随着Au负载量的升高或者降低,催化活性均有所降低.如,Au/GO-1催化剂对丁二烯的转化率为86%;Au/GO-0.2催化剂和Au/GO-2催化剂均表现出低催化活性,对丁二烯的转化率分别为3%和18%.图7(C)为催化加氢活性与纳米Au粒径的关系图.当Au纳米颗粒平均粒径为10.78 nm时,对1,3-丁二烯的催化活性仅为0.9×10-9molButadiene·s-1·g-1Au;纳米Au的粒径为15 nm时,催化活性约是10.78 nm的12倍,增大至12×10-9molButadiene·s-1·g-1Au;随后纳米Au粒径约17 nm时,催化活性下降至5×10-9molButadiene·s-1·g-1Au;当粒径达21 nm时催化活性大幅下降(0.5×10-9molButadiene·s-1·g-1Au).Nordlander等[38]研究表明,随着等离子体纳米颗粒粒径的增大,在光激发下产生的热电子数量增多,但热电子能量减小,反之亦然;而当粒径小于一定值(如小于10 nm)时,产生的热电子数量和能量均减小,这与纳米颗粒的态电子密度的离散性有关.最佳粒径时等离激元共振效应产生的有效高能热电子最多.由催化活性计算结果可得出,等离激元共振驱动的光热催化加氢反应存在最佳粒径(约15 nm),基于Au/GO的光转热催化选择性加氢测试可知,等离激元共振效应与Au纳米颗粒粒径大小直接相关[38~40],Au纳米颗粒粒径为15 nm时,具有非常好的光转热催化加氢性能.
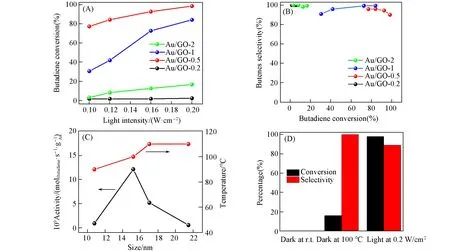
Fig.7 Photothermal hydrogenation performance of 1,3-butadiene over Au/GO catalyst with different light intensities(A,B),Au nanoparticles sizes(C)and comparison of catalytic hydrogenation properties of different reaction pathways for Au/GO-0.5(D)
根据光转热催化加氢测试结果,进一步研究了光转热驱动催化加氢反应的优势.选用Au/GO-0.5催化剂为模型催化剂,比较了传统热催化途径和本文的光转热驱动途径的催化加氢活性.由图7(D)可以看出,Au/GO-0.5催化剂在暗环境的室温下不表现出催化加氢活性;在暗环境中外部加热至100℃,反应90 min的热催化中对丁二烯的转化率为16%;而基于纳米金光照条件(0.2 W/cm2)下的等离激元共振光转热(100℃)驱动的催化反应,经90 min反应丁二烯的转化率为99%,约为暗环境下热催化性能的6倍.这表明等离激元共振光转热能大大增强负载纳米金的选择性催化加氢性能.针对这一结果,为了探索无光和有光条件下Au/GO-0.5催化剂是否对催化加氢活性有影响,对其在0.2 W/cm2光照下进行了原位XPS测试[图8(A)].结果表明,光照下Au/GO-0.5催化剂中负载的金属Au0会向Auδ+物种发生转变.这是由于光照下Au纳米颗粒表面发生了电子转移[41],从而产生了Au+(85.0,88.4 eV)和Au3+(86.1,89.4 eV),该物种在前期研究[42~44]中被证实是不饱和烯炔烃选择性催化加氢的高活性物种.Au/GO光电流响应测试证实,光照能促进电子在载体与负载金之间的转移[图8(B)].从[图8(B)]中可以看出,GO载体本身也会产生光生电子,这是因为GO上有大量缺陷存在而造成的[45],负载Au纳米颗粒后,GO载体的光生电子效应明显增强.在室温下打开氙灯,Au/GO-0.5催化剂产生的光电流最大,即产生的热电子最多[38],其它的依次为Au/GO-1,Au/GO-2和Au/GO-0.2.因此,光照条件下,等离激元共振产生的热(光转热)和催化活性物种(Auδ+)是实现负载金催化性能增强的本征原因.同时,光电流响应和原位XPS测试证明,Au在粒径为15 nm时产生的电子最多,并且提供了加氢活性位点,故表现出最佳的催化加氢性能.

Fig.8 Comparison of in situ XPS spectra under 0.2 W/cm2 light with conventional XPS spectra for Au/GO-0.5(A)and photocurrent response of Au/GO catalyst under 0.2 W/cm2 light(B)
催化稳定性是催化反应中判断催化剂性能最重要的参数之一.图9给出了在0.2 W/cm2光照下,光转热温度为100℃,对Au/GO-0.5催化剂稳定性的测试结果.如图9所示,当间歇式催化反应经96次循环(144 h)测试后,Au/GO-0.5催化剂的光热催化加氢反应的转化率和选择性均保持稳定,未出现下降趋势.其中,丁二烯转化率一直保持在98%以上,且始终保持高丁烯选择性(约90%).这主要归因于Au纳米颗粒与GO载体间的强相互作用,导致在催化加氢反应中相邻的纳米颗粒间不易发生团聚.同时,由于负载金纳米粒子的等离激元高频振动,产生的高温使得颗粒表面不易发生积碳失活[46],保持高稳定性.根据稳定性测试结果可以得出,Au/GO-0.5催化剂样品具有144 h以上的高催化加氢稳定性,具有很大的应用潜力.
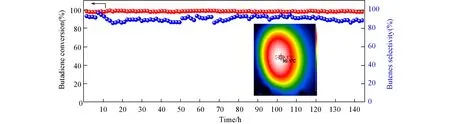
Fig.9 Stability test of Au/GO-0.5 catalyst under the light intensity of 0.2 W/cm2 in a batch reactor
3 结 论
通过阳离子吸附法,制备了氧化石墨烯负载的具有等离激元共振效应的纳米金催化材料(Au/GO),通过调变Au负载量(0.2%~2%),可控制备了粒径为10~21 nm的纳米金颗粒,并以工业单烯烃中常见丁二烯杂质的选择性催化加氢为探针反应,探究了金催化活性中心的等离激元共振光转热对金表面选择性催化加氢性能的增强机制.研究发现,在0.2 W/cm2光照条件下,随着金属负载量和粒径的增大,样品的光转热温度可升高至110℃,且光热转换效率高达88%;丁二烯转化率随Au负载量的增加先升高后降低,丁烯选择性在90%以上.当金负载量为0.5%(颗粒尺寸约为15 nm),光热转换温度为100℃,Au/GO-0.5表现出较高的丁二烯转化率(99%)和丁烯选择性(90%).催化剂经过144 h稳定性测试无失活趋势.相比同等条件的热催化反应,光转热驱动的Au/GO的催化活性提高了5倍.原位XPS测试发现,这种等离激元共振效应对选择性催化加氢性能增强的主要原因是,等离激元共振促进了催化活性物种(Auδ+)的生成,同时也提供热能来驱动催化加氢反应.本研究为工业不饱和烯炔烃的选择性催化加氢提供了一条绿色高效的反应路径.
猜你喜欢
能源工程(2022年2期)2022-05-23
东坡赤壁诗词(2022年1期)2022-02-25
科学导报(2021年81期)2021-11-27
烟台果树(2021年3期)2021-07-21
公民导刊(2018年11期)2018-05-14
农业与技术(2017年24期)2018-01-19
山西果树(2017年3期)2017-05-31
山东农业科学(2015年8期)2015-09-09
中学化学(2015年2期)2015-06-05
新课程·中学(2014年7期)2014-10-24
