
近六年中成药国家药品评价抽验微生物限度检查结果与分析
2022-10-13 09:10李玉立牛振东刘文杰井良义郝运伟张光华
首都食品与医药 2022年19期
李玉立,牛振东,刘文杰,井良义,郝运伟,张光华
(北京市药品检验研究院(北京市疫苗检验中心)国家药品监督管理局仿制药研究与评价重点实验室 中药成分分析与生物评价北京市重点实验室,北京 102206)
药品微生物限度检查于1972年在国内起步,卫生部1978年颁布了首个药品微生物限度标准,1995年版《中国药典》首次收载了微生物限度检查法,2000年版《中国药典》首次收载了微生物限度标准,2005年版《中国药典》强调了药品微生物检验中的“方法验证”,以避免因方法不合理而造成漏判、误判[1]。2015年版《中国药典》微生物标准体系实现了与国际标准的接轨,较好地解决了与ICH协调案标准的协调问题,且在推动药品微生物控制由“终产品检验向过程控制方向转变”方面取得了实质性进展[2]。
中成药在我国应用广泛,其主要采用中药材与中药饮片进行生产。2015年版《中国药典》收录了中药饮片品种理化性质分析的多种鉴别和检查方法,但相关微生物的具体检查方法和限度标准却长期处于缺失状态。2020年版《中国药典》[3]制剂通则要求,各种剂型的中成药的微生物限度均按照“非无菌产品微生物限度检查:微生物计数法”(通则1105)与“非无菌产品微生物限度检查:控制菌检查法”(通则1106)进行检查,同时2020年版《中国药典》(四部)增加了“中药饮片微生物限度检查法”(通则1108),但相关的微生物限度标准还不完善,而且对煎煮类中药饮片的微生物限度标准亦未作出统一、明确的规定[4]。
本文首先对近六年北京市药品检验研究院承担的中成药国家药品评价性抽验药品的微生物限度检查方法进行了验证和汇总,为以后的中成药国抽品种方法学适用性,甚至于该品种收入检验标准提供参考。同时,文中主要对所有品种微生物限度检查结果进行了汇总分析,在评价中成药卫生学质量的基础上,对中药饮片的微生物限度控制与其原料、生产工艺的相关性进行了进一步的分析,探讨了不同工艺所对应的微生物限度风险等级的不同,同时提及生产过程中可能存在的一些非传统工艺,例如射线辐照灭菌,为中成药微生物控制相关标准的完善及重点监管品种类型提供参考。
1 仪器、样品、菌株与培养基
1.1 仪器 LRH-250生化培养箱(上海一恒);电热脉动真空灭菌器(山东新华XG1.DMXD-0.36);生物安全柜(热电1300SERIESA2);PL2002电子天平(梅特勒);Max Q 6000恒温摇床(Thermo)。
1.2 样品 磁朱丸、板蓝根片、小金胶囊、小金丸、小金片、红金消结胶囊、红金消结片、柴黄胶囊、柴黄片、心脑欣片,均为近6年国家评价性抽验样品。
1.3 试验菌种 枯草芽孢杆菌Bacillus subtilis[CMCC(B)63501]、铜绿假单胞菌Pseudomonas aeruginosa [CMCC(B)10104]、金黄色葡萄球菌Staphylococcus aureus [CMCC(B)26003]、大肠埃希菌Escherichia coli[CMCC(B)44102]、白色念珠菌Candida albicans[CMCC(F)98001]、黑曲霉Aspergillus niger[CMCC(F)98003]、乙型副伤寒沙门菌Salmonella paratyphi B [CMCC(B)50094]。菌种均购自中国食品药品检定研究院,菌株传代数均为第Ⅲ代。
1.4 培养基 胰酪大豆胨液体培养基(TSB)、胰酪大豆胨琼脂培养基(TSA)、沙氏葡萄糖液体培养基(SDB)、沙氏葡萄糖琼脂培养基(SDA),由美国BD公司提供。pH7.0无菌氯化钠-蛋白胨缓冲液、麦康凯液体培养基、麦康凯琼脂培养基、肠道菌增菌液体培养基、紫红胆盐葡萄糖琼脂、RV沙门菌增菌液体培养基、木糖赖氨酸脱氧胆酸盐琼脂培养基,由北京奥博星生物技术有限责任公司提供。
2 方法与结果
2.1 方法 本次研究对不同厂家的样品按照《中国药典》通则1105、1106、1107进行了微生物限度检查方法适用性试验,最终确立微生物限度检查检验方法,具体如下。
2.1.1 磁朱丸 取本品10g,加胰酪大豆胨液体培养基至100ml,振摇、研磨至供试品分散均匀,制成1∶10的供试液。取1∶10的供试液1ml,加胰酪大豆胨液体培养基9ml,即为1∶100的供试液。同法10倍系列稀释至1∶1000的供试液。需氧菌总数测定,取1∶100的供试液1ml,注皿,依法检查(中国药典2020年版通则1105平皿法);霉菌和酵母菌总数测定,取1∶10的供试液1ml,注皿,依法检查(中国药典2020年版通则1105平皿法);耐胆盐革兰阴性菌检查,取1∶10的供试液10ml,置23℃培养2小时,按供试液制备方式,将预培养物稀释至1∶100和1∶1000的稀释液。取含10ml的肠道菌增菌液体培养基管3支,分别加入预培养后的1∶10、1∶100、1∶1000的稀释液各1ml,依法检查(中国药典2020年版通则1106);大肠埃希菌检查,取1∶10的供试液10ml,置胰酪大豆胨液体培养基100ml中,依法检查(中国药典2020年版通则1106);沙门菌检查,取本品10g,置胰酪大豆胨液体培养基200ml中,使供试品分散均匀,依法检查(中国药典2020年版通则1106)。
磁朱丸微生物限度标准为:需氧菌总数不得过105cfu/g,霉菌和酵母菌总数不得过5×102cfu/g,耐胆盐革兰阴性菌应小于102cfu/g,大肠埃希菌1g中不得检出,沙门菌10g中不得检出。
2.1.2 板蓝根片 取本品10g,加pH7.0无菌氯化钠-蛋白胨缓冲液至100ml,振摇至供试品分散均匀,制成1∶10的供试液。取1∶10的供试液1ml,加pH7.0无菌氯化钠-蛋白胨缓冲液9ml,即为1∶100的供试液。需氧菌总数测定,取1∶10的供试液1ml,注皿,依法检查(中国药典2020年版四部通则1105平皿法);霉菌和酵母菌总数测定,取1∶10的供试液1ml,注皿,依法检查(中国药典2020年版四部通则1105平皿法);大肠埃希菌检查,取1∶10的供试液10ml,置胰酪大豆胨液体培养基100ml中,依法检查(中国药典2020年版四部通则1106)。
板蓝根片微生物限度标准为:需氧菌总数不得过103cfu/g,霉菌和酵母菌总数不得过102cfu/g,大肠埃希菌1g中不得检出。
2.1.3 小金丸、小金胶囊、小金片 取本品10g,加胰酪大豆胨液体培养基至100ml,振摇,研磨至供试品分散均匀,制成1∶10的供试液。取1∶10的供试液1ml,加胰酪大豆胨液体培养基9ml,即为1∶100的供试液。取1∶100的供试液2ml,加胰酪大豆胨液体培养基8ml,即为1∶500的供试液。需氧菌总数测定,取1∶500的供试液1ml注皿,制备5份,合并计数,依法检查(中国药典2020年版通则1105平皿法);霉菌和酵母菌总数测定,取1∶10的供试液1ml注皿,依法检查(中国药典2020年版通则1105平皿法);耐胆盐革兰阴性菌检查,取1∶10的供试液10ml,置23℃培养2小时,按供试液制备方式,将预培养物稀释至1∶100和1∶1000的稀释液,取含10ml的肠道菌增菌液体培养基管3支,分别加入预培养后的1∶10、1∶100、1∶1000的稀释液各1ml,依法检查(中国药典2015版通则1106);大肠埃希菌检查,取1∶10的供试液10ml,置胰酪大豆胨液体培养基100ml中,依法检查(中国药典2020年版通则1106);沙门菌检查,取供试品10g,置胰酪大豆胨液体培养基200ml中,使供试品分散均匀,依法检查(中国药典2020年版通则1106)。
小金丸微生物限度标准为:需氧菌总数不得过3×104cfu/g,霉菌和酵母菌总数不得过102cfu/g,耐胆盐革兰阴性菌应小于102cfu/g,大肠埃希菌1g中不得检出,沙门菌10g中不得检出。
小金片、小金胶囊微生物限度标准相同,均为:需氧菌总数不得过104cfu/g,霉菌和酵母菌总数不得过102cfu/g,耐胆盐革兰阴性菌应小于102cfu/g,大肠埃希菌1g中不得检出,沙门菌10g中不得检出。
2.1.4 红金消结胶囊、红金消结片 取本品10g,加胰酪大豆胨液体培养基至100ml,使供试品分散均匀,制成1∶10的供试液。取1∶10的供试液10ml,置胰酪大豆胨液体培养基90ml中,即为1∶100的供试液。需氧菌总数测定,取1∶100的供试液1ml注皿,依法检查(中国药典2020年版通则1105平皿法);霉菌和酵母菌总数测定,取1∶10的供试液1ml注皿,依法检查(中国药典2020年版通则1105平皿法);耐胆盐革兰阴性菌检查,取1∶10的供试液10ml,置23℃培养2小时,按供试液制备方式,将预培养物稀释至1∶100和1∶1000的稀释液。取含10ml的肠道菌增菌液体培养基管3支,分别加入预培养后的1∶10、1∶100、1∶1000的稀释液各1ml,依法检查(中国药典2020年版通则1106);大肠埃希菌检查,取1∶10的供试液10ml,置胰酪大豆胨液体培养基100ml中,依法检查(中国药典2020年版通则1106);沙门菌检查,取本品10g,置胰酪大豆胨液体培养基200ml中,使供试品分散均匀,依法检查(中国药典2020年版通则1106)。
红金消结胶囊与红金消结片标准相同,均为:需氧菌总数不得过104cfu/g,霉菌和酵母菌总数不得过102cfu/g,耐胆盐革兰阴性菌应小于102cfu/g,大肠埃希菌1g中不得检出,沙门菌10g中不得检出。
2.1.5 柴黄片、柴黄胶囊 取本品10g,加胰酪大豆胨液体培养基至100ml,振摇至供试品分散均匀,制成1∶10的供试液。取1∶10的供试液1ml,加胰酪大豆胨液体培养基9ml,即为1∶100的供试液;取1∶100的供试液1ml,加胰酪大豆胨液体培养基9ml,即为1∶1000的供试液。需氧菌总数测定,取1∶10的供试液1ml注皿,依法检查(中国药典2020年版通则1105平皿法);霉菌和酵母菌总数测定,取1∶10的供试液1ml注皿,依法检查(中国药典2020年版通则1105平皿法);耐胆盐革兰阴性菌检查,取1∶10的供试液10ml,置23℃培养2小时。按供试液制备方式,将预培养物稀释至1∶100和1∶1000的稀释液。取含10ml的肠道菌增菌液体培养基管3支,分别加入预培养后的1∶10、1∶100、1∶1000的稀释液各1ml,依法检查(中国药典2020年版通则1106);大肠埃希菌检查,取1∶10的供试液10ml,置胰酪大豆胨液体培养基100ml中,依法检查(中国药典2020年版通则1106);沙门菌检查,取本品10g,置含3%聚山梨酯80和0.3%卵磷脂的胰酪大豆胨液体培养基1000ml(柴黄胶囊培养基量为500ml)中,使供试品分散均匀,依法检查(中国药典2020年版通则1106)。
柴黄胶囊和柴黄片标准相同,均为:需氧菌总数不得过104cfu/g,霉菌和酵母菌总数不得过102cfu/g,耐胆盐革兰阴性菌应小于102cfu/g,大肠埃希菌1g中不得检出,沙门菌10g中不得检出。
2.1.6 心脑欣片 取本品10g,加胰酪大豆胨液体培养基至100ml,振摇至供试品分散均匀,制成1∶10的供试液。取1∶10的供试液1ml,加胰酪大豆胨液体培养基9ml,即为1∶100的供试液。需氧菌总数测定,取1∶100的供试液1ml注皿,依法检查(中国药典2020年版通则1105平皿法);霉菌和酵母菌总数测定,取1∶10的供试液1ml注皿,依法检查(中国药典2020年版通则1105平皿法);耐胆盐革兰阴性菌检查,取1∶10的供试液置23℃培养2小时,按供试液制备方式,将预培养物稀释至1∶100和1∶1000的稀释液。取含10ml的肠道菌增菌液体培养基管3支,分别加入预培养后的1∶10、1∶100、1∶1000的稀释液各1ml,依法检查(中国药典2020年版通则1106);大肠埃希菌检查,取1∶10的供试液10ml,置胰酪大豆胨液体培养基100ml中,依法检查(中国药典2020年版通则1106)。沙门菌检查,取本品10g,置胰酪大豆胨液体培养基500ml中,使供试品分散均匀,依法检查(中国药典2020年版通则1106)。
心脑欣片标准为:需氧菌总数不得过104cfu/g,霉菌和酵母菌总数不得过102cfu/g,耐胆盐革兰阴性菌应小于102cfu/g,大肠埃希菌1g中不得检出,沙门菌10g中不得检出。
2.2 结果与分析 选取历年抽验比例最高的三家企业,以“品名+A/B/C”表示。所示数据为近六年国抽部分中成药微生物限度检查结果,包括需氧菌总数、霉菌和酵母菌总数、耐胆盐革兰阴性菌、沙门菌、大肠埃希菌。样品涉及6类中成药的10个品种,共计662批次,涵盖了丸剂、片剂及胶囊剂等不同剂型,包括如原粉入药、煮提浓缩等不同生产工艺。通过对其微生物学指标检查结果的统计分析,可见近年整体微生物限度合格率较高,600余批次的检测中仅有2批次需氧菌总数不合格,1批次霉菌和酵母菌总数不合格,整体风险可控。见表1。
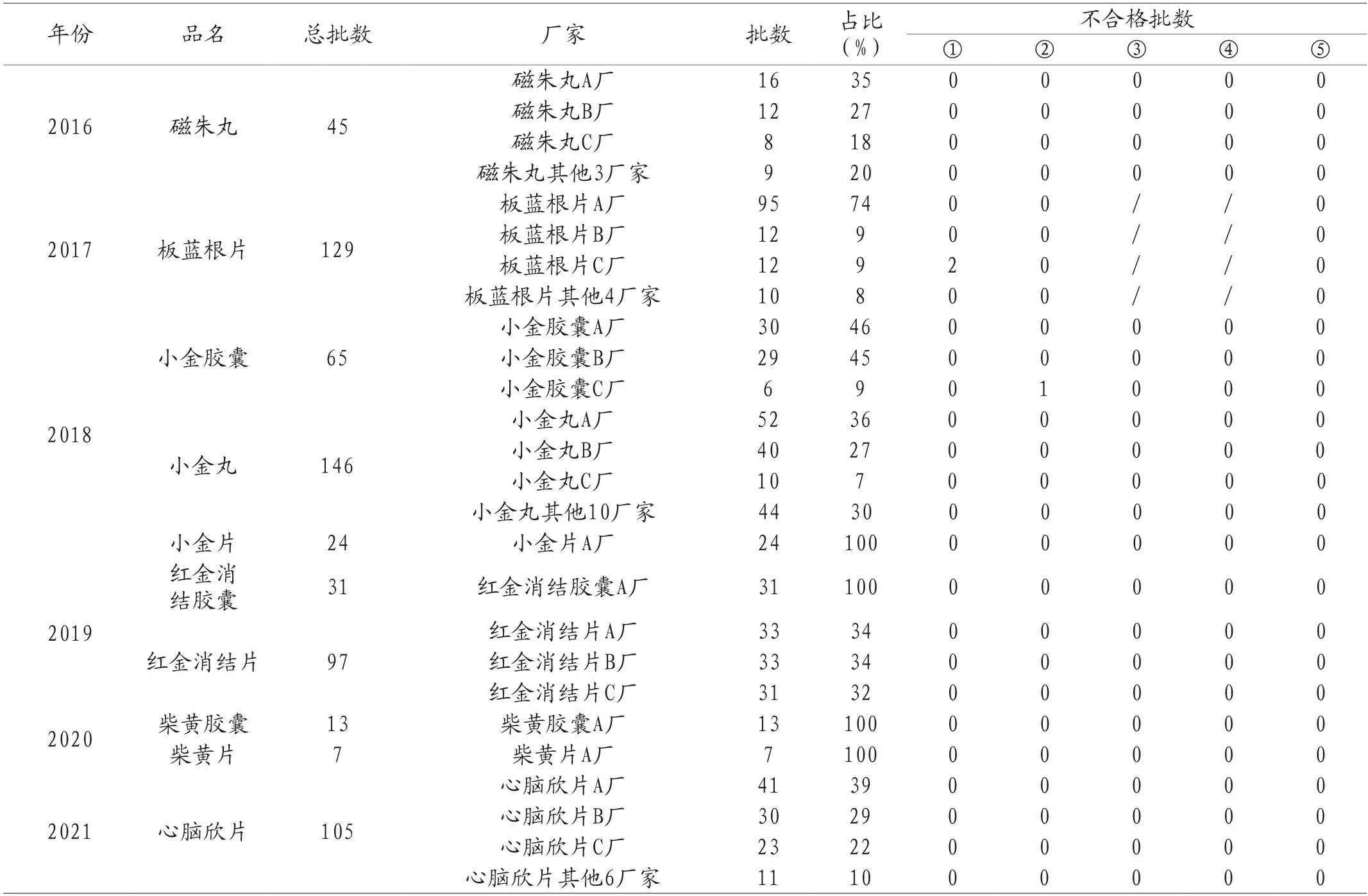
表1 近六年中成药国家药品评价抽验微生物限度检查结果
以下将对历年结果分别进行汇总分析,探讨各个品种的总体情况及可能存在的问题。
2016年国家药品评价性抽验品种为磁朱丸,45批次样品分别来自河北龙海药业有限公司等6个厂家,合格率100%。磁朱丸由磁石(煅)、朱砂和六神曲(炒)三味药组成,经由原料药粉末配研后制备成丸,该品种批准生产的剂型均为水丸。所有批次产品的微生物限度检测结果如表2所示。
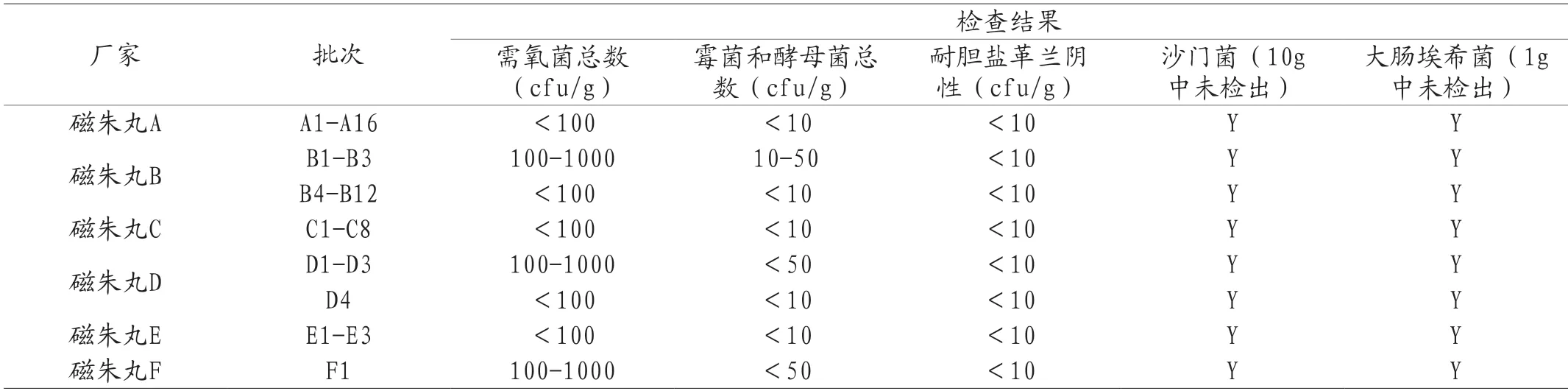
表2 2016年磁朱丸国家药品评价抽验微生物限度检查结果
由于磁朱丸制备工艺中涉及发酵类原粉入药,限度标准较宽,所有检验结果的菌落数指标都远低于标准,且不同厂家批次之间差异不大,82%的样品在试验中无菌落生长,卫生学总体质量稳定。
2017年国家药品评价性抽验品种为板蓝根片,129批样品分别来自大理白族自治州中药制药有限公司等7个厂家。总合格率98.4%,有两批次需氧菌总数严重超标,均来自C厂家。板蓝根片以板蓝根为原材料,经水煮醇提后浓缩制备而成,剂型为糖衣片剂。所有批次产品的微生物限度检查结果如表3所示。
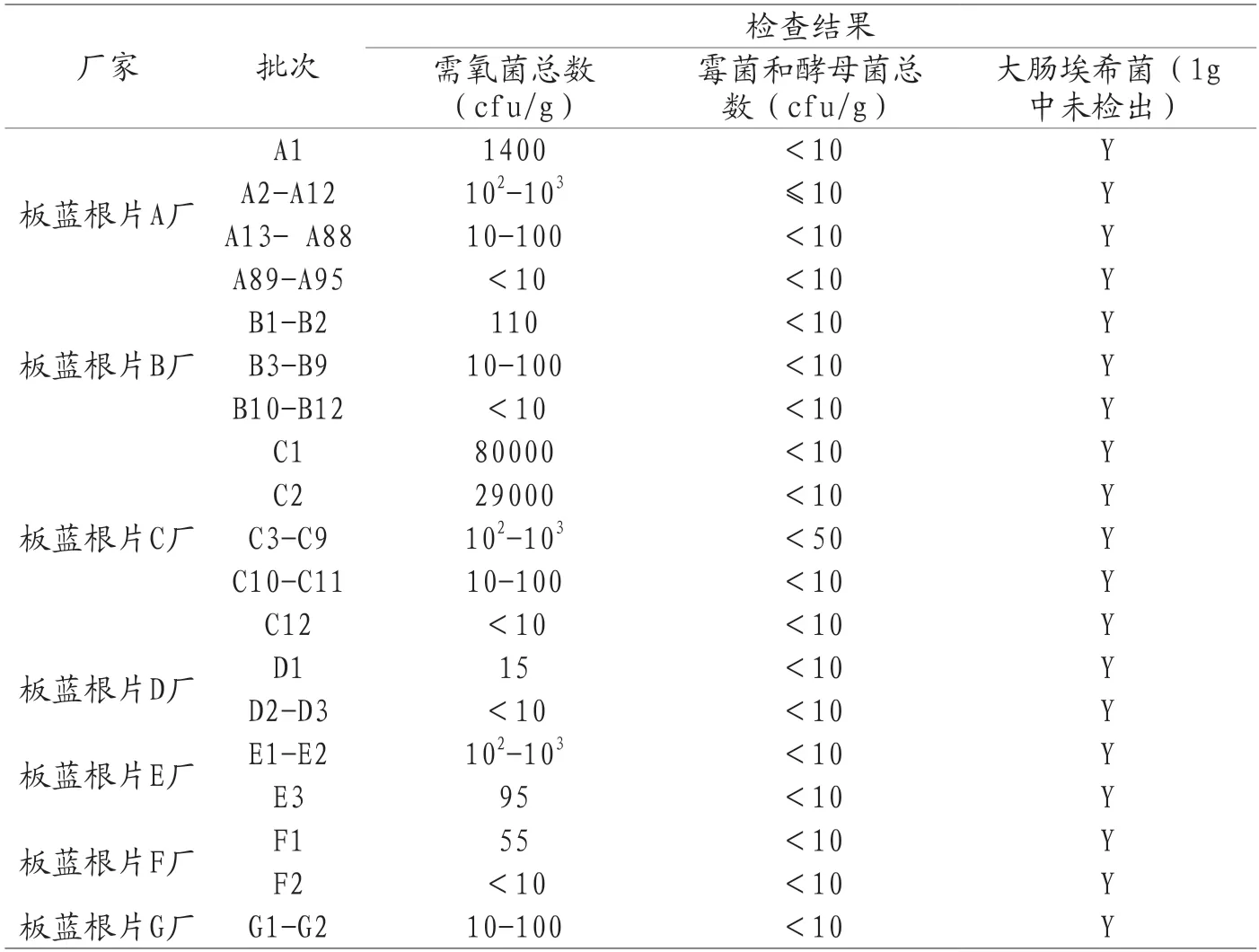
表3 2017年板蓝根片国家药品评价抽验微生物限度检查结果
板蓝根片制备工艺是一个全提取的过程,“水煮醇提”的工艺相当于对原料药进行了一次“除菌”。在此前提下,板蓝根片C厂家有两批次样品需氧菌总数分别达到了80000cfu/g和29000cfu/g,远远超出了103cfu/g的合格标准。且该厂家被抽验的12批次样品中含菌量整体都超出其他厂家,说明该厂家生产过程中的微生物控制存在较大问题,不同批次控制程度差异较大,提示存在随机性的生产质控风险,已提示该企业及时查找问题,加强微生物污染风险管控。板蓝根片A厂家被抽验批次数最多,占比达74%,且整体含菌量基本一致。除板蓝根片C厂家以外,其他厂家的微生物限度检查合格率均为100%。特别提示,在合格的基础上,大多数批次的检验过程中都有菌落生长,说明“煎煮提取”的工艺可在一定程度上降低微生物负载,但难以完全杀灭耐热微生物(如芽孢杆菌、霉菌和酵母菌孢子等)[5]。并且,如生产环境和关键工艺微生物污染控制不足还可能带来外源性微生物污染,影响终产品的卫生质量。
2018年国家药品评价性抽验品种有小金丸146批次,合格率100%;小金胶囊65批次,合格率98.5%,有1批霉菌和酵母菌总数超标,来自厂家C;小金片24批次,合格率100%。
小金丸146批样品分别来自北京同仁堂股份有限公司同仁堂制药厂等13家企业。小金丸由人工麝香、制草乌、地龙等十味药制成,制备工艺中涉及原粉入药。所有批次产品的微生物限度检查结果如表4所示。146批样品均符合规定,B厂家的40批样品与H厂家的4批样品的工艺稳定性可进一步加强控制。
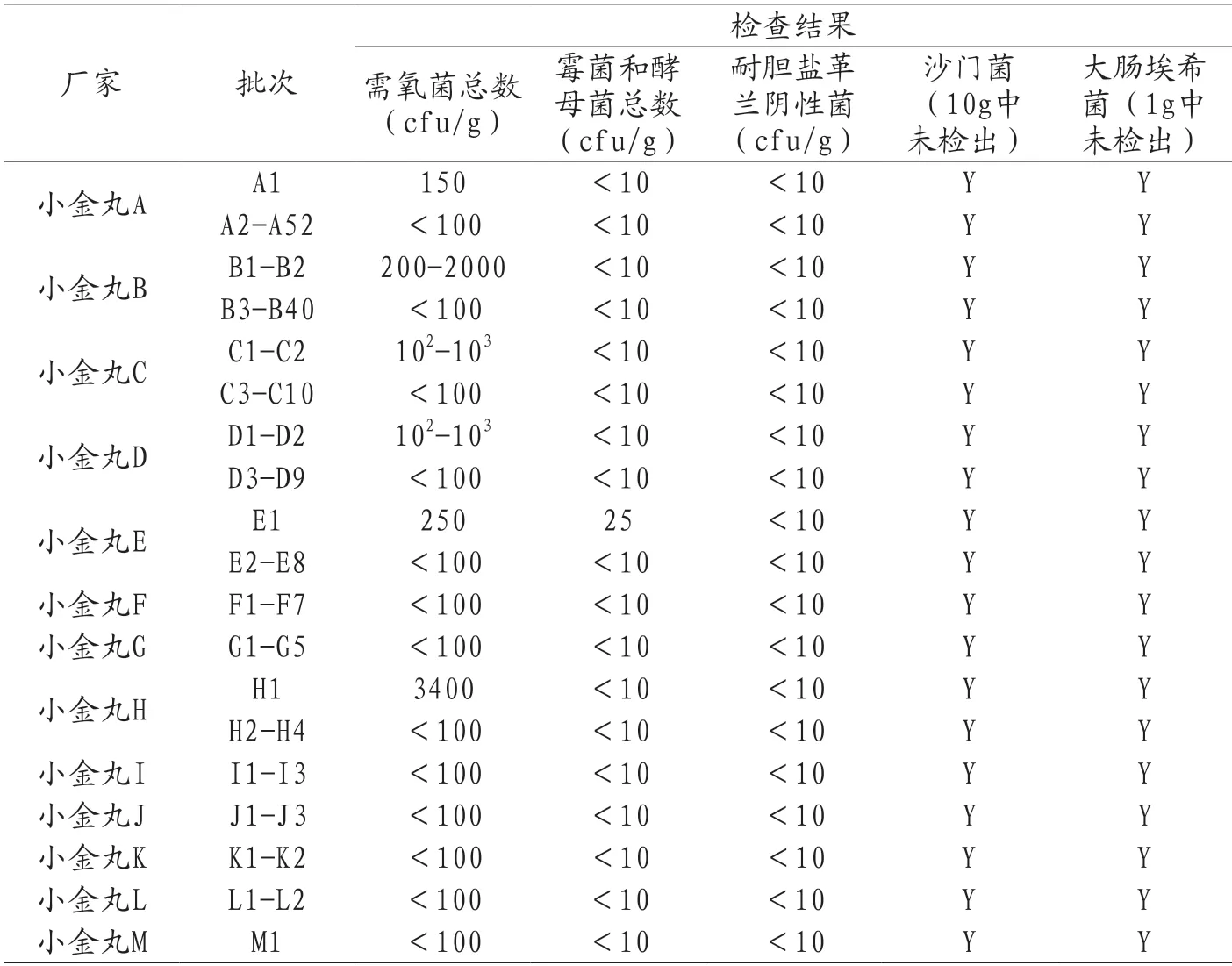
表4 2018年小金丸国家药品评价抽验微生物限度检查结果
小金胶囊65批样品分别来自健民药业集团股份有限公司等3家企业。检测结果详见表5,64批结果均符合规定,厂家C的1批产品霉菌和酵母菌总数超标。
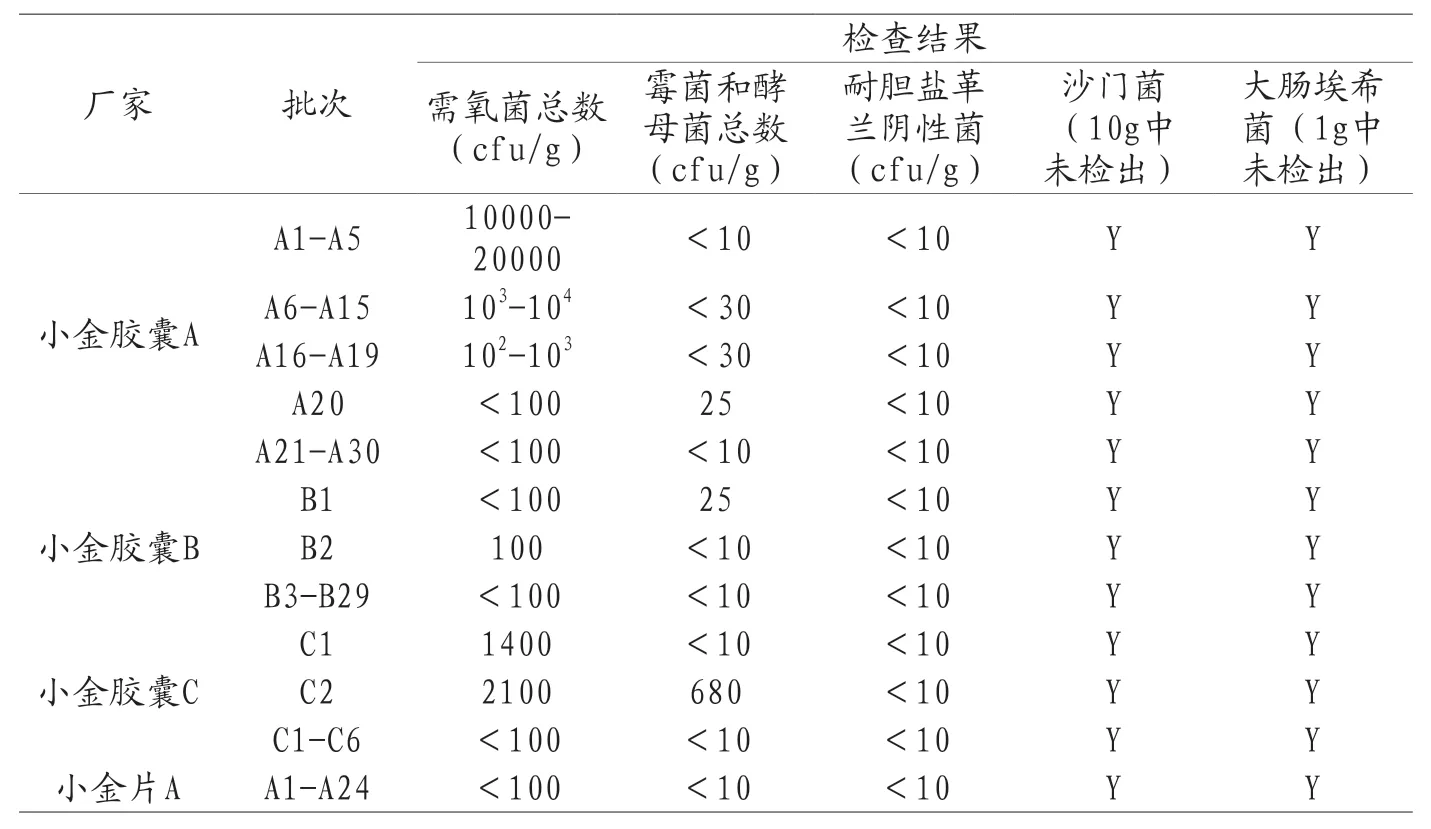
表5 2018年小金胶囊和小金片国家药品评价抽验微生物限度检查结果
小金片24批样品全部来自小金片A企业。检测结果详见表5,24批结果均符合规定。所有批次产品的微生物限度检查结果如表5所示。
小金丸、小金胶囊与小金片三个品种处方一致,剂型不同,均包含原粉入药,小金丸的需氧菌总数限度要求较小金胶囊与小金片更宽松。三个品种检测中的含菌量存在一定差异,小金片所有批次均无菌落生长,小金丸大多批次无菌落生长,个别有菌落生长的批次其含量也远低于限度要求。而小金胶囊不同厂家的检测结果存在差异,涉及的三个厂家中小金胶囊B厂家的检测结果趋势与小金片、小金丸类似,C厂家有1批产品霉菌和酵母菌总数超标,超标结果为680cfu/g;厂家C共抽样6批样品,其余5批霉菌和酵母菌结果为<10cfu/g,提示该厂家微生物污染的生产控制存在较大偶发性风险,应及时进行风险查找并严格管控。A厂家的大多数批次都有菌落生长,且含量不低,有些批次接近上限,在104cfu/g的水平上。结合该品种的处方工艺,A厂家的生产过程需要加强批次稳定性的控制。
2019年国家药品评价性抽验品种有红金消结胶囊31批次,合格率100%,样品全部来自红金消结胶囊A企业;红金消结片97批次,合格率100%,样品来自山东绿因药业有限公司等三个厂家。
红金消结胶囊由三七、柴胡和黑蚂蚁等十味药制成,其中五味研粉后进行灭菌备用,另五味煎煮后制备稠膏,然后混合制成胶囊剂。红金消结片与红金消结胶囊处方相同,工艺类似,最终将灭菌后的原粉和煎煮制备的稠膏混合物制成片剂。所有批次产品的微生物学指标检测结果如表6所示。
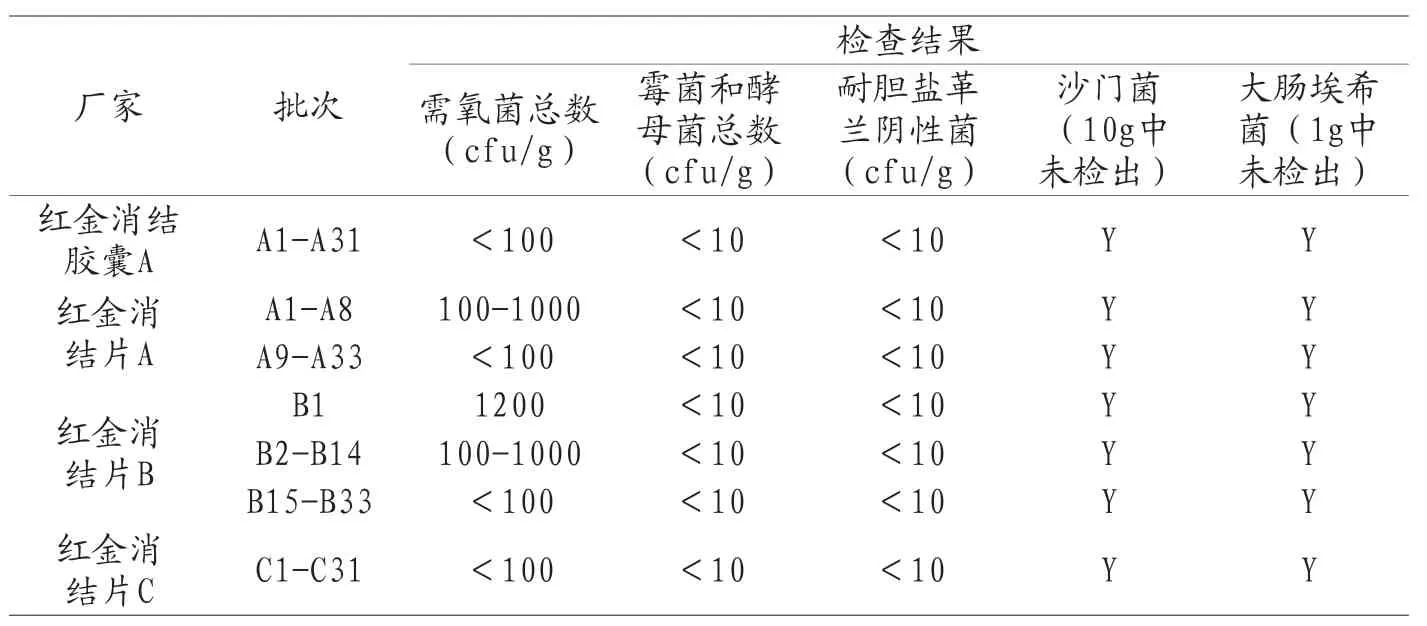
表6 2019年红金消结胶囊及红金消结片国家药品评价抽验微生物限度检查结果
红金消结胶囊与红金消结片虽然均有原粉直接入药,但其制备过程中明确说明了原粉经过了灭菌工艺。其限度检测结果总体趋势一致,大多批次无菌落生长,个别有菌落生长的样品其含量大概在102cfu/g水平,远低于限度要求。
2020年国家药品评价性抽验品种有柴黄胶囊13批次,合格率100%,样品来自柴黄胶囊A企业;柴黄片7批次,合格率100%,样品来自柴黄片A企业。
柴黄胶囊和柴黄片处方相同,由柴胡和黄芩两味药制备而成。柴黄胶囊制备工艺中将药材原粉灭菌后使用,柴黄片则是药材原粉直接入药。所有批次产品的微生物学指标检测结果如表7所示。
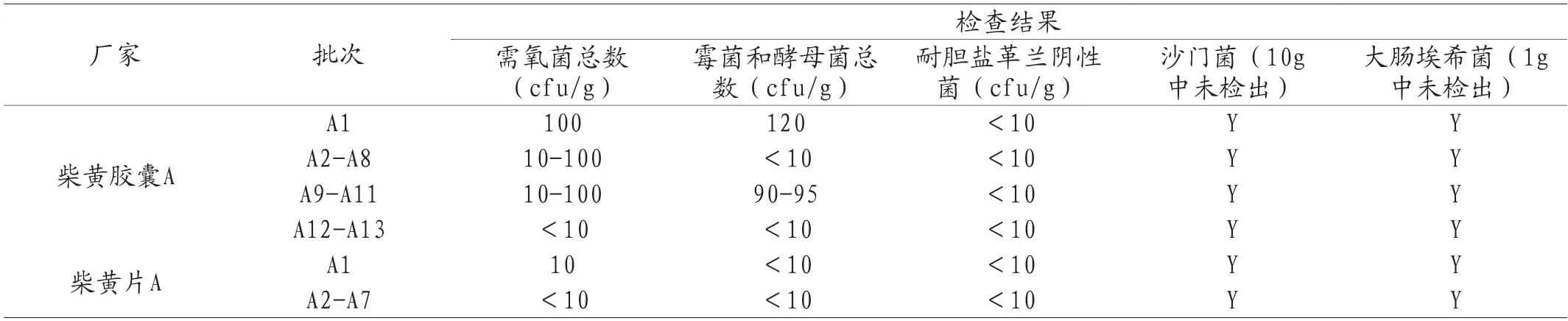
表7 2020年柴黄胶囊及柴黄片国家药品评价抽验微生物限度检查结果
柴黄胶囊及柴黄片处方一致,剂型不同且二者制备工艺有较大差别,柴黄胶囊制备中涉及了原料的“预除菌”,而柴黄片没有,二者的检测结果却相反,柴黄胶囊大多批次有菌落生长,而柴黄片绝大多数无菌落生长。
2021年国家药品评价性抽验品种心脑欣片105批次,合格率100%,样品来自北京御生堂制药有限公司等9个厂家。
心脑欣片由红景天、枸杞子和沙棘鲜浆三味药制备而成。制备工艺中红景天醇提、枸杞子水煮后入药,沙棘鲜浆在50℃浓缩后入药。所有批次产品的微生物学指标检测结果如表8所示。
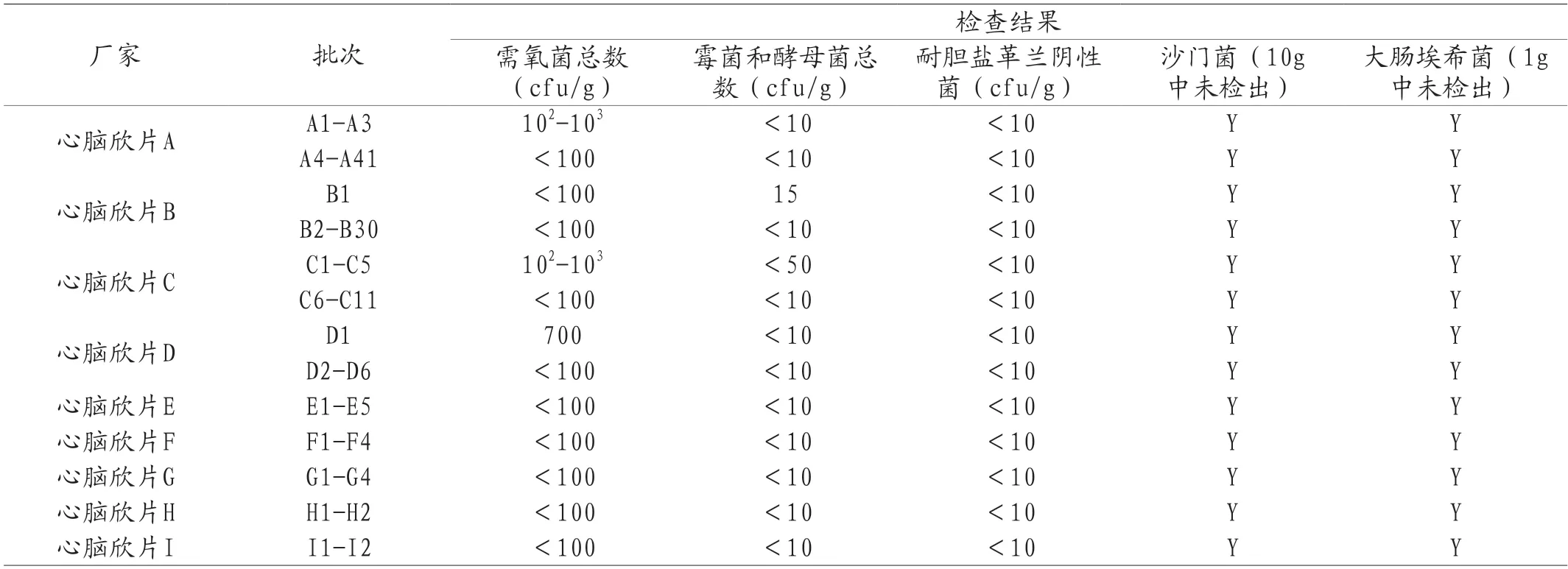
表8 2021年心脑欣片国家药品评价抽验微生物限度检查结果
根据心脑欣片的检验结果,其整体含菌量远低于标准,且批次间差异不大,整体质量较为稳定。50℃的原浆入药也会起到一定的“除菌”作用,一定程度上会降低微生物载量。
3 讨论
3.1 近六年中成药国家药品评价抽验微生物限度检查方法学整体情况分析 近六年样品整体涵盖了中成药的不同剂型及不同生产工艺,本文中针对各个品种进行方法学验证试验并进行了分析,从结果来看,大多数中成药的抑菌作用不是很强,对抑菌作用强的品种,菌落总数检查可采用高稀释级供试液,而控制菌检查除了增加增菌液的体积外,还可以在增菌液中添加3%聚山梨酯80和0.3%卵磷脂,方法具有很强的参考意义,可为以后的中成药微生物限度检查方法学适用性提供参考。
3.2 近六年中成药国家药品评价抽验微生物限度检验结果整体情况分析 微生物限度的控制是保障口服制剂用药安全的核心。中成药物料和生产工艺都具有一定特殊性。
中成药来源主要是中药材,包括植物、动物类内脏、尸体及某些矿物质。这些中药材自身携带大量的微生物和虫卵,特别是对那些有生药粉直接入药的制剂微生物污染风险较高[6]。生产过程中的水飞、煮提、醇提、浓缩等工艺,包括辅料的加入,都可能带来微生物污染。
通过对历年数据的比对可见,在合格率很高的前提下,药物实际含菌量与标准相差很大,远低于标准所规定的限度。一般药品生产中原粉直接入药的情况下,成品应与饮片原粉的含菌量在相似的水平上。结合文献中的一些数据[7-10],饮片原粉的含菌量是较高的,而药物实际含菌量,包括需氧菌总数、霉菌和酵母菌总数远低于标准。例如,2020年抽验品种柴黄片,黄芩原粉直接入药的情况下,其微生物载量却低于黄芩原粉灭菌后再入药的柴黄胶囊,其可能原因将在3.3中进一步探讨。
结合历年品种,对于有原粉或原浆直接入药的品种而言,其微生物载量应该较高,而经历的“水煮醇提”“原粉灭菌”之类工艺的品种其微生物超标可能性较小。
3.3 近六年中成药国家药品评价抽验品种生产工艺对微生物限度结果的影响 本文中全提取的药物和原粉入药的药物,检验结果没有明显差别,甚至部分品种中原粉入药的含菌量更低。其在制备中应该是引入了非传统工艺的一些灭菌方式。另有一些品种,都是原粉入药,但有些工艺中明确提及原粉是在灭菌后入药的。此外,还有一些品种虽未提到灭菌的过程,但其最终的菌落计数结果与前者相当,分析其工艺中也引入了类似的灭菌步骤。
例如,2017年的板蓝根片经过了“水煮醇提”的除菌过程,但是检验结果中绝大多数批次在合格的基础上有菌落的生长,需氧菌含量在10-102cfu/g居多,而例如“小金丸”这种含有大量菌群的饮片原粉直接入药的品种,反而大多数批次的检验中无任何菌落生长。
再如红金消结片和红金消结胶囊虽然也涉及原粉入药,其制备过程明确指出了会将原粉灭菌后备用,但是相比其他原粉入药且制备中无灭菌工艺的药物,微生物的载量水平却差异不大,甚至更高。从这样的结果趋势可以看出,其他品种的药物在生产过程中或者“终产品”也经历过类似的“灭菌过程”。结合现有的一些文献[11-17],可以了解到,包括辐射、蒸汽灭菌、微波灭菌等都是会用到的方式。有一些文献表明,经历这些非传统工艺后药物质量无明显变化,但是缺乏更明确的对质量无影响的研究论证。目前业界也正在开展非传统除菌工艺对质量影响的课题研究,为下一步更好的指导中成药的微生物限度控制提供更严谨的技术支撑和方法指导[18-19]。
3.4 本文中所有品种的微生物限度检查方法针对每个厂家都进行了方法学验证,如能将文中所列品种的微生物限度检查方法收入到该品种质量标准微生物限度检查项下,可明显方便检验。
猜你喜欢
文萃报·周五版(2020年24期)2020-06-22
中国药房(2018年3期)2018-10-19
中学教学参考·理科版(2017年8期)2018-02-24
中国质量万里行(2017年10期)2017-11-08
新校园·中旬刊(2017年5期)2017-07-25
新高考·高二数学(2016年11期)2017-07-06
人民周刊(2016年11期)2016-06-30
新高考·高二数学(2015年11期)2015-12-23
发明与创新·中学生(2015年5期)2015-06-08
数理化学习·初中版(2009年3期)2009-04-21
