
16S rRNA gene sequencing analysis on changes in the intestinal f lora of Procambarus clarkii with “Black May”disease*
2022-10-08 01:16JieGONGGuoqingSHENMengruZHUMingZHANChangjunXIYanSHUIZenghongXUHuaishunSHEN
Jie GONG , Guoqing SHEN , Mengru ZHU , Ming ZHAN , Changjun XI ,Yan SHUI , Zenghong XU , Huaishun SHEN ,**
1 Wuxi Fisheries College, Nanjing Agricultural University, Nanjing 210095, China
2 Key Laboratory of Freshwater Fisheries and Germplasm Resources Utilization, Ministry of Agriculture, Freshwater Fisheries Research Center, Chinese Academy of Fishery Sciences, Wuxi 214081, China
Abstract The morbidity and mortality peak of farmed Procambarus clarkii occurs around May every year, a phenomenon known as “Black May” disease (BMD). Increasing evidence shows that the intestinal f lora is closely related to host health. We analyzed and compared the microbiota of healthy and BMDaff ected P. clarkii intestines. The results show that there was no signif icant diff erence in bacterial α-diversity(richness P=0.59; evenness P=0.43; and diversity P=0.052) between the diseased group and the control group. Four dominant phyla in the intestines of crayf ish in the control group, namely Tenericutes (30.86%),Bacteroidetes (29.99%), Firmicutes (22.23%), and Proteobacteria (15.23%), were identif ied. However, a striking shift in the microbial composition were found in the intestines of P. clarkii with BMD. Bacteroidetes was a dominant phylum in healthy P. clarkii, whereas the prevalence was low in diseased P. clarkii (1.87%).By contrast, the prevalence of Proteobacteria was signif icantly higher ( P <0.05) in P. clarkii with BMD than in P. clarkii without BMD. Candidatus Bacilloplasma, Bacteroides, Vibrio, and Aeromonas showed signif icant diff erences ( P <0.05) at the genus level. Tax4Fun function prediction indicated that the relative abundance of genes involved in energy metabolism in the intestinal f lora of P. clarkii with BMD was signif icantly reduced ( P <0.05). Therefore, BMD can change the composition of the intestinal microbiota of P. clarkii. This study contributes to the understanding of the relationship between intestinal f lora and host especially in aquatic animals.
Keyword: Procambarus clarkii; “Black May” disease (BMD); intestinal f lora; high-throughput sequencing
1 INTRODUCTION
Convincing evidence suggests that the intestinal microbiota plays an important role in maintaining host health, including nutrient absorption, pathogen defense, and antibiotic resistance (Ott et al., 2004;Sekirov et al., 2010; Dai et al., 2017). In addition, the intestinal microbial f lora, which produces signaling molecules, can inf luence host metabolism, such as short-chain fatty acid (SCFA) production and vitamin synthesis (Watanabe et al., 2006). Therefore, the balance of the intestinal ecosystem is very important for host health, and a change in bacterial diversity may lead to host disease (Guarner, 2007; Othman et al., 2008). The outbreak of diseases in aquaculture is usually accompanied by disorder of the intestinal f lora. Therefore, characterizing the diff erences in intestinal f lora between healthy and diseased hosts can be a f irst step in improving disease prevention and treatment (Berry et al., 2012; Berry and Reinisch,2013). Studies have explored the diff erences in intestinal microf lora between healthy and diseased aquatic animals. InPenaeusvannameiwith acute hepatopancreatic necrosis disease (AHPND),Vibriowas enriched in the intestine (Dong et al., 2021).There were signif icant diff erences in intestinal microf lora between healthy and diseased (suff ering from furunculosis)Coreiusguichenoti(Li et al.,2016). Some studies have shown that changes in the intestinal bacterial community are closely related to the severity ofP.vannamei, and the indicator group can be used for health assessment (Xiong et al., 2015).However, the analysis of crustacean intestinal microbiota is still in its infancy, and it is not clear how diseases aff ect the intestinal microbiota.
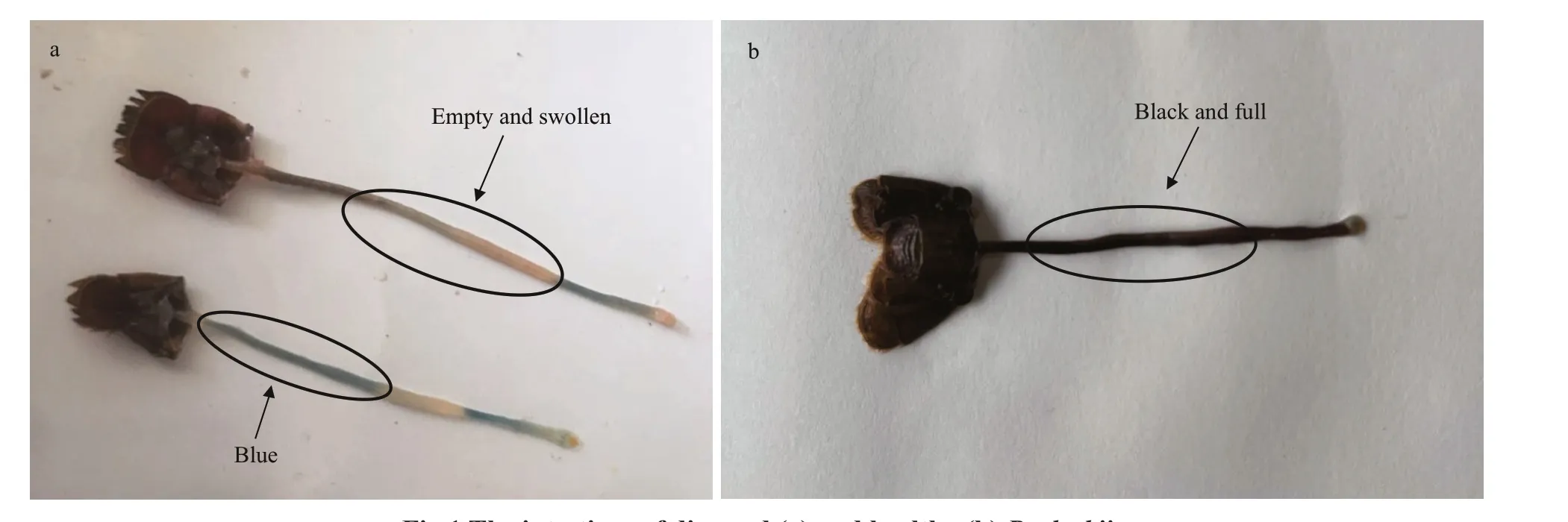
Fig.1 The intestines of diseased (a) and healthy (b) P. clarkii
Procambarusclarkii, a species of crayf ish, is classif ied into arthropods, crustaceans, Decapoda,and the family Labridae.P.clarkiiis native to northern Mexico and the southern USA, and it has spread to Europe, Africa, Asia, and other parts of the Americas(Gherardi, 2006). The species was introduced to China in 1929 (Yue et al., 2008). In recent years, it has become an important species in the aquaculture industry due to its high protein, rich nutrition, and delicious f lavor. However, the peak of the disease season typically occurs in May, during the farming of crayf ish, and the phenomenon occurred in May is commonly known as “Black May” disease (BMD)(Huang et al., 2020). Diseased crayf ish usually exhibit reduced food intake. Their carapace is easily peeled off and movement is slow. Dissection of diseased individuals reveals that the intestine is empty, blue,and swollen (Fig.1a). BMD has caused tremendous economic losses to the crayf ish farming industry.Once a pond becomes aff ected, nearly 90% mortality would be typically reported. At present, there are few studies on BMD inP.clarkiiin China, so little is known about how BMD aff ectsP.clarkii. Considering that the intestines ofP.clarkiiwith BMD are empty and swollen, it is necessary to characterize the changes in intestinal microbiota.
To explore the inf luence of BMD on the intestinal f lora ofP.clarkii, we compared the intestinal f lora of healthy and BMD-diseasedP.clarkii, and found associated changes in the intestinal f lora. The results provide a theoretical reference for the study of the intestinal bacteria ofP.clarkiiand will help in the prevention and treatment of this disease.
2 MATERIAL AND METHOD
2.1 Sampling
The diseased crayf ish used in the experiment were collected from aP.clarkiifarm with BMD in Xuyi County, Jiangsu Province, China. The organisms exhibited reduced food intake and lethargy. Dissection of diseased individuals reveals that the intestine is empty, blue, and swollen. Our samples (control and diseased) were collected from diff erent ponds. The intestines of 10 healthy (control) and 10 diseasedP.clarkiiwere collected, frozen in liquid nitrogen, and stored at -80 ℃. And our laboratory through PCR amplif ication and observation with transmission electron microscopy to detect the pathogens in the sampled crayf ish, including white spot syndrome virus (WSSV) andProcambarusclarkiidicistro-like virus (PcDV, a novel Dicistro-like virus discovered inProcambarusclarkiiwith “Black May”disease). In the end, both pathogens were positive (Huang et al.,2020).
2.2 Extraction of genomic DNA and PCR amplif ication of 16S rDNA
The total genomic DNA ofP.clarkiiintestine was extracted by the cetyltrimethylammonium bromide(CTAB) method, and the concentration of isolated DNA was measured with a NanoDrop 2000 spectrophotometer (GE Healthcare, USA). The samples were diluted to 1 ng/μL with sterile water and used to amplify the 16S rDNA of intestinal microorganisms.
The diluted genomic DNA was used as the template for PCR amplif ication. PCR amplif ication of the 16S rRNA gene was performed using PCR primers (515F-806R) specif ic for the V3-V4 regions. The primer sequences were as follows: 5′-GTGCCAGCMGCCGCGGTAA-3′/5′-GGACTACHVGGGTWTCTAAT-3′. PCR products were detected by electrophoresis with 2% agarose gels. Then, PCR products were purif ied with GeneJET Gel Extraction Kit (Thermo Scientif ic). Before sequencing, a spectrophotometer was used to determine the DNA concentration of each PCR product, and the DNA was mixed in an appropriate ratio according to the sequencing requirements.
2.3 High-throughput sequencing
Twenty samples were sequenced in a commercial company (Novogene, Beijing), of which 10 samples were from the control group and 10 were from the diseased group. Sequencing libraries were generated using an Illumina TruSeq DNA PCR-Free Library Preparation Kit (Illumina, USA) following the manufacturer’s recommendations and index adapters were added. The library quality was assessed on the Qubit 2.0 Fluorometer (Thermo Scientif ic) and Agilent Bioanalyzer 2100 system. Finally, the library was sequenced on an Illumina Nova Seq platform and 250-bp paired-end reads were generated.
2.4 Sequence analysis
Paired-end reads were assigned to samples based on their unique barcode and truncated by cutting offthe barcode and primer sequence. Paired-end reads were merged using FLASH (V1.2.7, http://ccb.jhu.edu/software/FLASH/) to obtain the Raw tags (the spliced tags sequence) (Magoč and Salzberg, 2011).According to the tag quality control process of Qiime(V1.9.1, http://qiime.org/scripts/split_libraries_fastq.html) (Caporaso et al., 2010), the following operations were carried out: tags interception: truncate raw tags from the f irst low-quality base site with continuous low-quality value (the default quality threshold is≤19) and the base number reaches the set length (the default length is 3); tags length f iltering: f ilter out tags with continuous high-quality base length less than 75% of tags length. Eff ective tags were obtained through read splicing, quality control, and removal of chimeric sequences (https://github.com/torognes/vsearch/) (Rognes et al., 2016).
2.5 Statistical method
To study the species composition of each sample,the eff ective tags of all samples were clustered into operational taxonomic units (OTUs) with 97%identity (Uparse v7.0.1001, http://www.drive5.com/uparse/) (Haas et al., 2011). The OTU sequences were taxonomically annotated by Mothur and SILVA132(http://www.arb-silva.de/). According to the OTU clustering results, a Venn diagram was drawn to show common and unique OTUs between diff erent groups(http://bioinformatics.psb.ugent.be/webtools/Venn/).According to the species annotation results, the relative abundances of species at diff erent taxonomic levels were plotted.
The evaluation of microbial diversity in a natural environment involves two aspects: species richness and evenness information. Alpha diversity was used to analyze the richness and diversity of the withincommunity microbial community, as shown by rank abundance. Alpha diversity indices (Good’s coverage,the Chao1 estimator, the ACE, the Simpson index,and the Shannon index) were calculated in QIIME(Version 1.9.1) to analyze the abundance and diversity of microbial communities in the two groups. R(version 2.15.3) was used to analyze the diff erence of alpha diversity index between groups. Beta diversity was used to analyze the microbial community composition of diff erent samples. QIIME was used to calculate Unweighted UniFrac distance and construct unweighted pair group method with arithmetic(UPGMA) sample clustering tree. Principal coordinate analysis (PCoA) was conducted by the WGCNA packages, STAT packages, and GGPLOT2 packages of R (Version 2.15.3), based on Unweighted UniFrac distance. Moreover, linear discriminant analysis(LDA) eff ect size (LEfSe) was used to identify biomarkers with signif icant diff erences between the groups (the default f ilter value of the LDA score is 4).
The 16S rRNA gene sequences of prokaryotes in the kyoto encyclopedia of genes and genomes(KEGG) database were extracted and compared to those in the SILVA SSU Ref NR database (BLAST bitcore >1 500) by the BLASTN algorithm to establish the correlation matrix. The KEGG database prokaryotic whole genome function information annotated by UProC and PAUDA was mapped to the SILVA database to realize the functional annotation of the SILVA database.
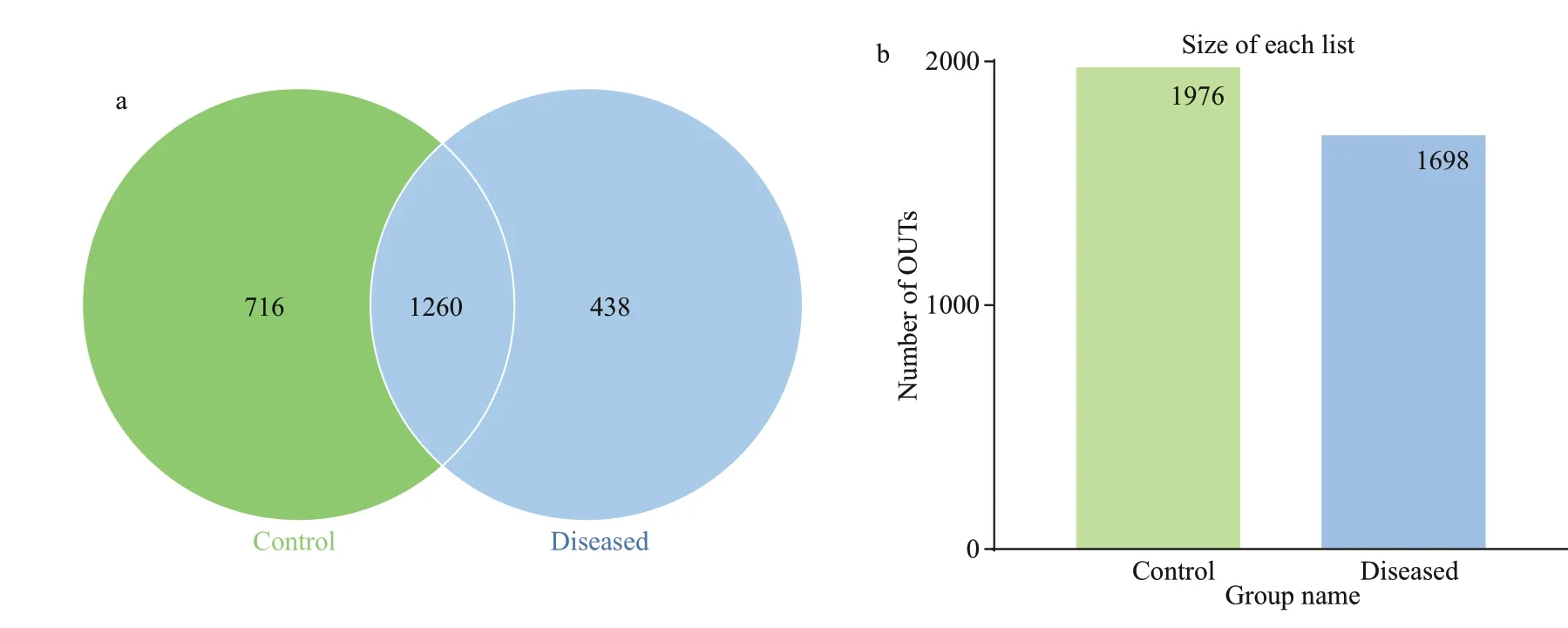
Fig.2 The Venn diagrams (a) and the number of OTUs in control and diseased group (b)
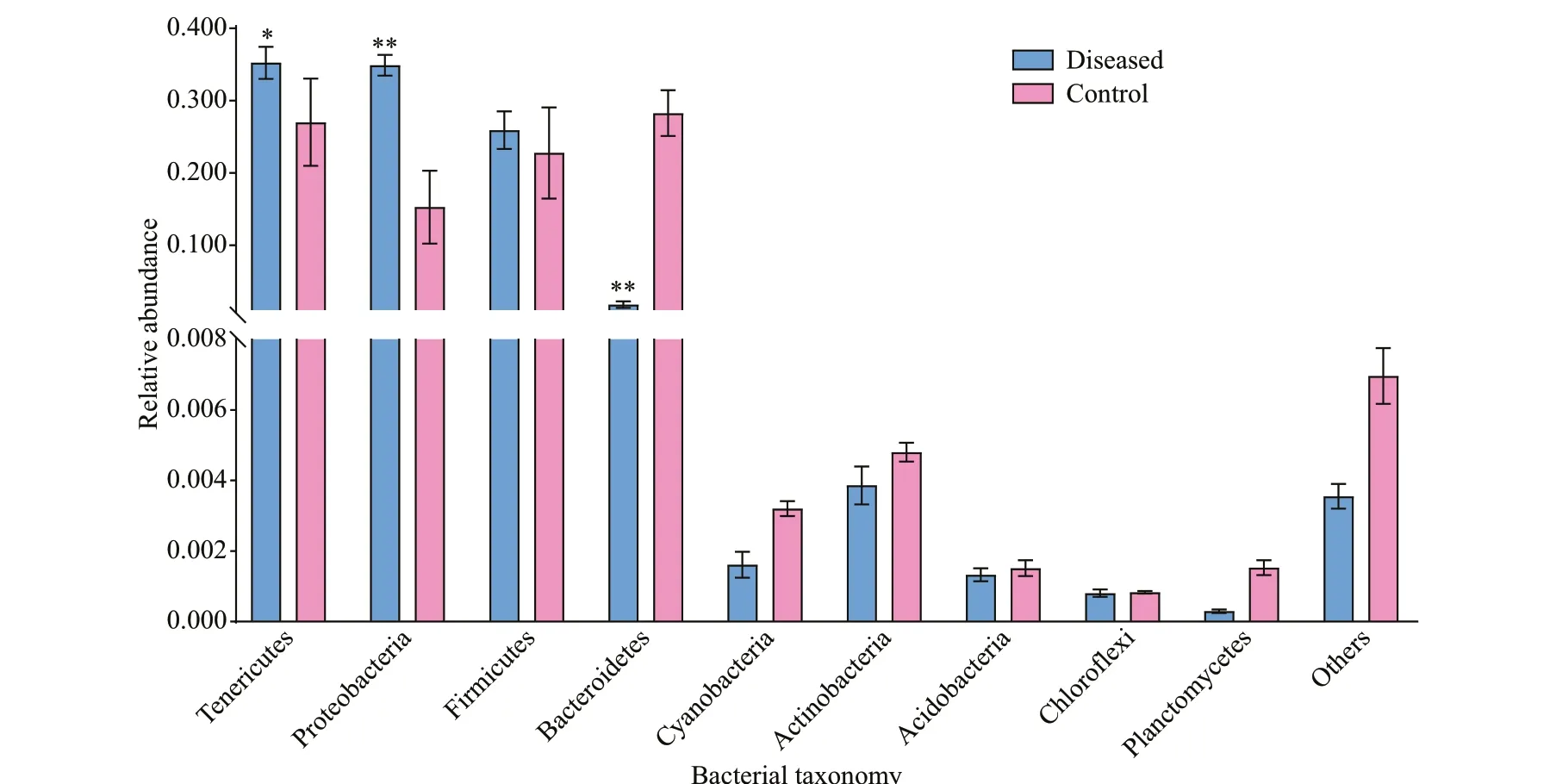
Fig.3 Abundance of intestinal microf lora of P. clarkii at the phylum level in the control and diseased groups
3 RESULT
3.1 The high-throughput sequencing data analysis
In total, through the splicing of reads, 85 772 tags per sample were measured, and 82 002 valid data were obtained through quality control, with the amount of eff ective data reaching 77 703 tags after removing chimaera sequences. With 97% identity, the sequences were grouped into OTUs, with 2 432 OTUs. In the Venn diagram, the two circles represent the diseased group and the control group, and the numbers represent the number of OTUs. Among them, 1 260 OTUs were shared between the two groups, 438 OTUs were unique to the diseased group,and 716 OTUs were unique to the control group(Fig.2).
3.2 The intestinal f lora composition
In this study, 47 phyla were recognized in the intestinal f lora of control and diseased groups. As shown in Fig.3, we identif ied four dominant phyla in the intestines of crayf ish in the control group, namely Tenericutes (30.86%), Bacteroidetes (29.99%),Firmicutes (22.23%), and Proteobacteria (15.23%).We also identif ied three dominant phyla in the diseased group, namely, Tenericutes (35.71%),Proteobacteria (34.59%), and Firmicutes (26.65%).Although Tenericutes had the highest abundance in both groups, and there was no signif icant diff erence(P>0.05), a striking shift in the microbial composition was found in the intestines ofP.clarkiiwith BMD.Bacteroidetes was a dominant phylum in healthyP.clarkii, whereas its prevalence was low in diseasedP.clarkii(1.87%). By contrast, the prevalence of Proteobacteria was signif icantly higher (P<0.05) inP.clarkiiwith BMD than inP.clarkiiwithout BMD.The percentage of bacteria belonging to the phylum Proteobacteria in diseased crayf ish was higher than that in healthy crayf ish, which was mainly due to the contributions of Alphaproteobacteria (P<0.05). In contrast, the percentage of Bacteroides in diseased crayf ish was signif icantly lower than that in healthy crayf ish (P<0.05).
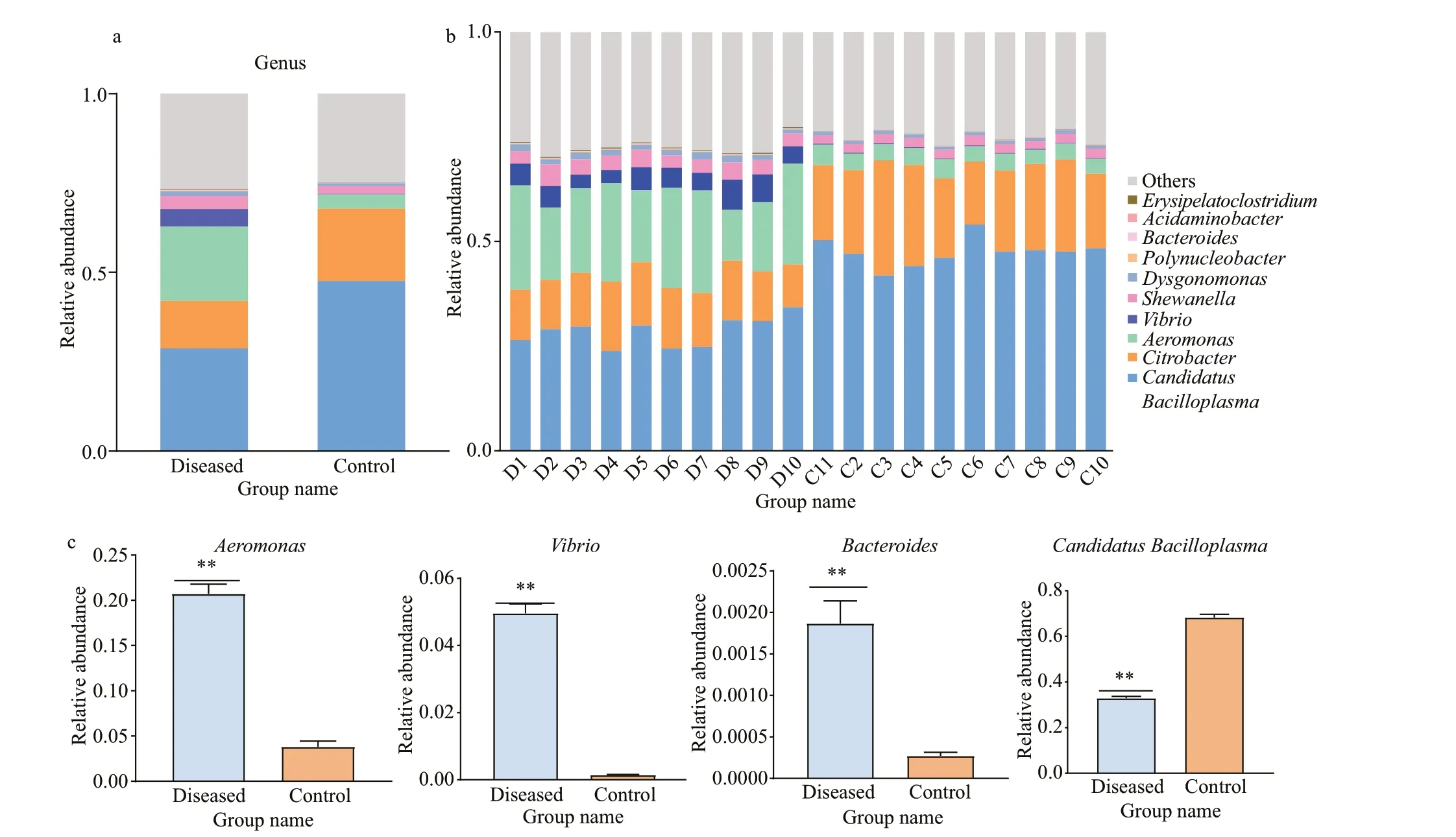
Fig.4 The relative abundances of the intestinal bacterial community among the diseased and control samples at the genus level
As shown in Fig.4a, the composition of the intestinal f lora ofP.clarkiishowed more relative diff erences at the genus level. In the annotation results, a total of 1 150 (47.29%) OTUs annotations were at the genus level. Among them, the dominant intestinal bacteria ofP.clarkiiin the control group wereCandidatusBacilloplasma(47.56%)andCitrobacter(20.30%). The dominant intestinal bacteria in the diseased group wereCandidatusBacilloplasma(28.72%),Citrobacter(13.30%), andAeromonas(20.76%). AlthoughCandidatusBacilloplasmawas the dominant intestinal bacterium in both the control group and the diseased group, its abundance was signif icantly lower (P<0.01) in the diseasedd group. In addition, other genera with signif icant enrichment in the diseased group wereAeromonas(P<0.01) andVibrio(P<0.01) (Fig.4c).The individual relative abundances for samples in the control and diseased group were shown in Fig.4b, and the detailed data were shown in Supplementary Tables S1 & S2.
3.3 Diversity analysis
The results of alpha diversity analysis show that the Simpson, ACE, Chao1, and Shannon indices in the diseased group were slightly lower than those in the control group (Supplementary Table S3), but there was no signif icant diff erence in alpha diversity indices between the groups (richnessP=0.59; evennessP=0.43; and diversityP=0.052) (Fig.5a).
PCoA of all 20 samples showed that the clustering of sample points in the control group and the diseased group were relatively close, indicating that the diff erence in intestinal f lora among individuals within each group was small. In contrast, the clusters of sample points of the control group and the diseased group were far apart, indicating a certain diff erence between intestinal f lora groups (Fig.5b). PCoA indicated that PC1, which ref lected the bacterial community, could separate the healthy group from the diseased group, and the total contribution of other principal components was 15.08%.
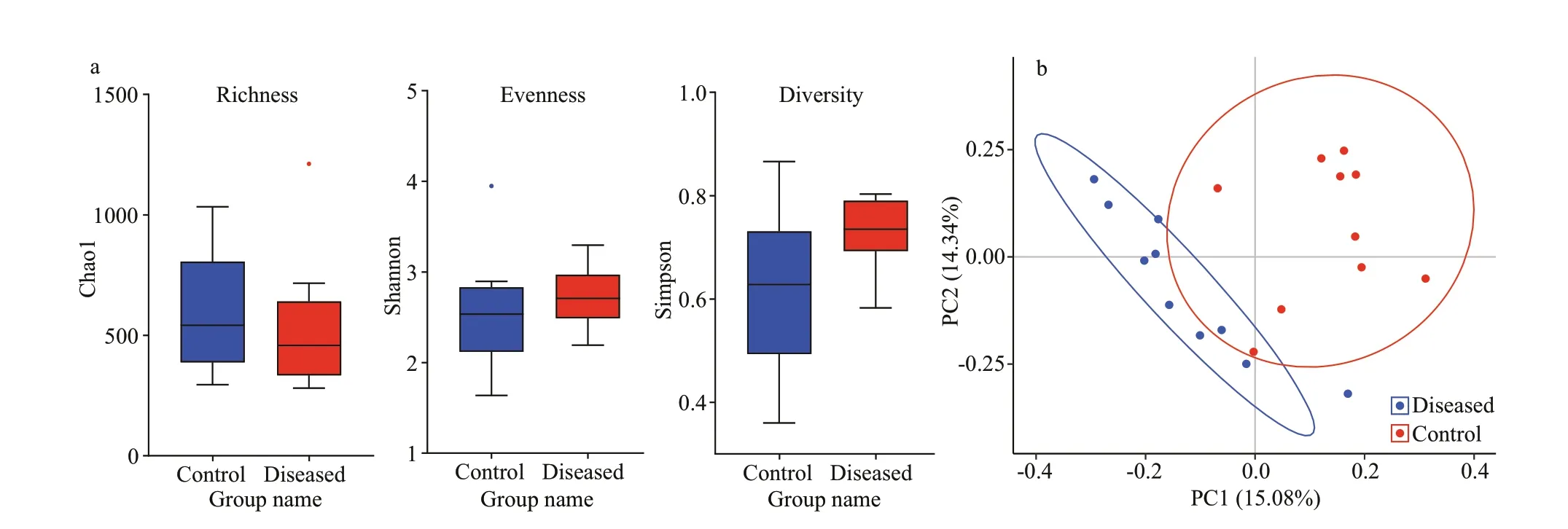
Fig.5 Measures of α-biodiversity, including richness (Chao1), evenness (Shannon), and diversity (Simpson) (a), and principal component analysis (PCA) (b)
In addition, a multi response permutation procedure (MRPP, based on Bray-Curtis distance)analysis was performed to examine the diff erences in intestinal microbiota between the two groups, as shown in Table 1. The diff erence between groups was expressed as the expected-delta, which was greater than the diff erence within groups(signif icance=0.031).
To further analyze the intestinal f lora ofP.clarkiirelated to BMD, LEfSe was used to compare the relative abundance of intestinal f lora between the control group and the diseased group (Fig.6a). There were 5 signif icantly diff erent f lora constituents between the control group and the diseased group,belonging to three phyla: Cyanobacteria, Firmicutes,and Proteobacteria. Among them, we found that the bacteria of the diseased group showed a signif icant increase in the abundances of Enterobacteriaceae and Vibrionaceae of Proteobacteria. Their relationship is shown in Fig.6b.
3.4 16S rRNA functional prediction analysis
To analyze the functional changes in intestinal microbiota inP.clarkiiwith BMD, Tax4Fun was used to predict the metagenomic potential between the two groups. The results show that the high abundance of bacterial metagenomes in the intestine of healthy and BMD-diseasedP.clarkiiwas mainly related to the metabolic pathway group, genetic information processing group and environmental information processing group at KEGG level 1. ThereThe smaller the observed-delta value, the smaller the diff erence within the group; the larger the expected-delta value, the greater the diff erence between the groups. TheR-value was greater than 0, indicating signif icant diff erences between groups.Pvalue less than 0.05 indicates a signif icant diff erence.was no signif icant diff erence between the KEGG level 1 microbial function of the diseased group and the control group (P>0.05). For the metabolism group, the largest abundance was found in amino acid metabolism, carbohydrate metabolism, and energy metabolism in level 2. For the genetic information processing group, the highest abundance was found in replication and repair and translation. In the environmental information processing group, the greatest abundance was observed in the membrane transport pathway. The specif ic data analysis of the above results is shown in Supplementary Fig.S1.Specif ically, the microbial functions related to replication and repair in diseased group were signif icantly more abundant (P<0.05) than those in the control group. More detailed changes were observed at KEGG Level 2. As shown in Fig.7, the microbial functions related to Glycan biosynthesis and metabolism and Enzyme families in the diseased group were signif icantly more enriched (P<0.05) than those in the control group, while those assigned to the Amino acid metabolism, Energy metabolism, and Metabolism of cofactors and vitamins were signif icantly more enriched (P<0.05) in the control group than in the diseased group.

Table 1 MRPP and Adonis analysis of the control group and diseased group (based on Bray-Curtis distance)
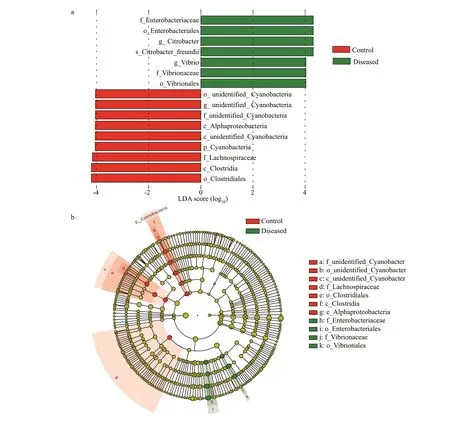
Fig.6 Linear discriminant analysis (LDA) (a) and Cladogram analysis (b)
4 DISCUSSION
Procambarusclarkiiis widely cultivated in China because of its strong environmental adaptability and great commercial value. As the scale of production has increased, outbreaks of disease inP.clarkiihave occurred frequently, especially around May, when the incidence rate and mortality rate ofP.clarkiireach their peak, phenomenon known as “Black May”disease (BMD). At present, there are many controversies about the cause of BMD. It is generally considered that BMD inP.clarkiiis caused by large f luctuations in temperature and water temperature in May. WSSV and some Vibrio species reproduce in large numbers and cause proliferative diseases inP.clarkii(Chen, 2019). BMD is also believed to be caused by the accumulation of ammonia nitrogen and nitrite due to the rapid rise of water temperature and low dissolved oxygen in the water (Shen et al., 2020).According to the symptoms of the empty, swollen intestines ofP.clarkiiwith BMD, the characteristics and structure of intestinal bacteria in crayf ish with BMD and healthy crayf ish were determined by 16S rRNA gene sequencing.
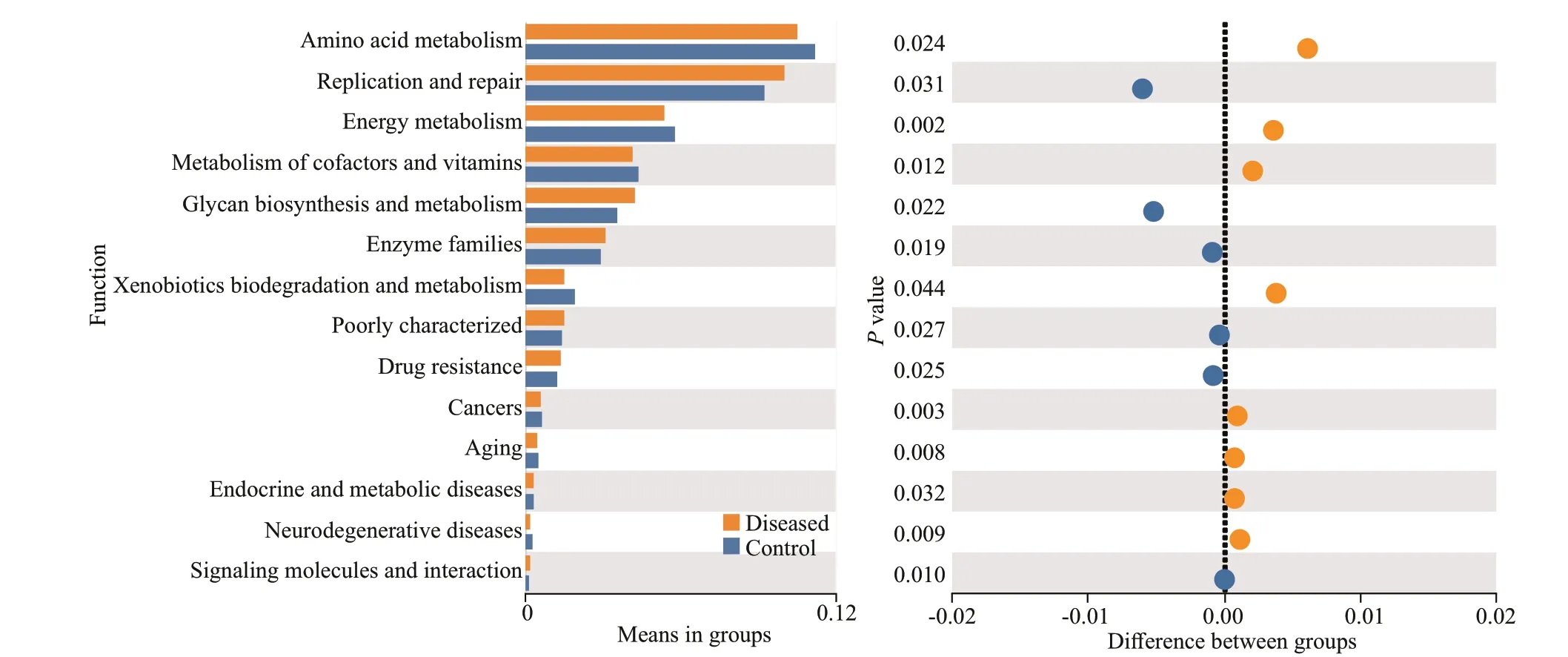
Fig.7 Extended error bar chart to determine the signif icant diff erence (95% conf idence interval) between the average proportion of functional genes in the control (blue) and diseased (orange) samples
Crustaceans, such asP.clarkii, have a large number of microorganisms inhabiting their intestines. These microorganisms are the products of long-term coevolution with the host (Serrano-Villar et al., 2016). We identif ied four dominant phyla,namely, Proteobacteria, Bacteroidetes, Firmicutes,and Tenericutes, in the intestines of healthy crayf ish,which conf irms a previous study on the dominance of these taxa in the intestinal bacterial community ofP.clarkii(Liu et al., 2020a; Zhang et al., 2020,2021). In the present study, the alpha diversity of the bacteria in the intestinal tract ofP.clarkiidid not diff er signif icantly (P>0.05) between control and diseased groups. However, there was a signif icant diff erence in the prevalence of the main bacterial phyla in the intestine of the groups with and without BMD, which changed the composition of the intestinal f lora. We found that compared with healthy crayf ish, there was a signif icant enrichment of Proteobacteria (P<0.05) in the intestines of diseased crayf ish (Fig.3). In contrast,Bacteroideswas prevalent in the intestines of healthy crayf ish, while its prevalence was low in diseased crayf ish. As the most abundant bacteria in f ishponds, Proteobacteria participates in various biogeochemical processes in aquatic ecosystems (such as carbon, nitrogen, and sulfur cycles) (Klase et al., 2019; Li et al., 2021).Studies have shown that Proteobacteria may be essential for certain physiological and biochemical functions of crustacean intestines, such asPortunustrituberculatus(Zeng et al., 2016; Li et al., 2018).And an increased abundance of Proteobacteria was a potential risk of disease inL.vannamei(Fan and Li,2019).Bacteroidesmay be related to intestinal fat storage, and the nutrient absorption ofBacteroidesis negatively correlated with the presence of fat deposits (Xu et al., 2007; Guo et al., 2008). The balance of the intestinal microbial population is very important to the health of the host, and a loss or increase in intestinal microbial diversity can lead to host diseases (Semova et al., 2012; Li et al., 2021).The enrichment of Proteobacteria abundance and the decrease inBacteroidesindicate that BMD may aff ect the fat metabolism and nutrient absorption function of the intestinal microbiota ofP.clarkii. In addition, it was reported that Alphaproteobacteria may cause local infection of tissue inLitopenaeusvannamei(Huang et al., 2016). Alphaproteobacteria are enriched in the intestines of diseased crayf ish(P<0.05). However, further studies are needed to determine whether Alphaproteobacteria is related to damage to the intestinal tissues ofP.clarkiiwith BMD.
At the genus level, the diff erence in the abundance of the intestinal f lora ofP.clarkiibetween the control group and the diseased group was mainly observed inCandidatusBacilloplasma(P<0.01),Bacteroides(P<0.01),Aeromonas(P<0.01) andVibrio(P<0.01)(Fig.4c).CandidatusBacilloplasmais considered to be a novel lineage of Mollicutes associated with the hindgut wall of the terrestrial isopodPorcellioscaber(Crustacea: Isopoda) and was f irst discovered inPorcellioscaber(Kostanjš ek et al., 2007). The enrichment ofCandidatusBacilloplasmawas also previously found in the intestine ofP.vannamei(Hou et al., 2018), suggesting thatCandidatusBacilloplasmais prevalent in the intestines of crustaceans. Members of the genusBacteroidesare necessary to break down complex molecules in the intestines and help the body’s immune system f ight against potentially harmful pathogens (Gregory et al., 2015). In addition,Bacteroidesis important for maintaining intestinal homeostasis, especially through the production of short-chain fatty acids (SCFA) (Zhong et al., 2015).Moreover, studies have shown thatBacteroidesis rich in enzymes related to carbohydrate transport and protein metabolism, which are essential processes of digestion (Karlsson et al., 2011; Zhou and Zhi, 2016).The over-enrichment ofBacteroidesmay be related to the regulation of the homeostasis of the intestinal f lora byP.clarkii. The Tax4Fun function prediction further supports the view that the abundance of genes involved in metabolism (Energy metabolism and Metabolism of cofactors and vitamans) in the intestinal f lora ofP.clarkiiwith BMD is signif icantly lower (P<0.05)than that of controlP.clarkii(Fig.7), which indicates that energy metabolism is disrupted by BMD. This also further explains the symptoms of the jejunum and reduced food intake ofP.clarkiiwith BMD.
In the intestine ofP.clarkiiin BMD, we detected signif icant upregulation of some opportunistic pathogens:Vibrio(0.152%-4.939%,P<0.01) andAeromonas(from 3.965% to 20.758%,P<0.01)(Fig.4c). Generally, many members of theVibriogenus are considered the main pathogens causing disease and death in aquatic animals. For example,Vibrioparahaemolyticuscan cause AHPND and “gill rot disease” in shrimp and crayf ish (Alloul et al., 2021).Studies onP.clarkiishow that pathogenic bacteria such asVibriomay be an opportunistic factor for disease (Shui et al., 2020). When the host’s immunity decreases or the number of bacteria exceeds the normal value, host disease can occur (Hou et al., 2018).Aeromonashydrophila, a more common conditional pathogen, exists widely in aquatic environments. Wang et al. (2004) noted that pathogenicAeromonashydrophilacan cause many diseases, such as tremor and oedema, inEriocheirsinensis, and it is also one of the main bacterial pathogens in shrimp and crab farming operation. Although conditional pathogenic bacteria are signif icantly enriched in the intestine of crayf ish with BMD, further studies are needed to determine whether the two are related to the pathogenesis of BMD in crayf ish.
In the present research, the functional prediction of the intestinal f lora indicated that the metagenomic potential of the intestinal f lora ofP.clarkiihas a higher abundance in the Carbohydrates metabolism and Amino and acids, which may be attributed to the carbohydrates and proteins from the host being the main energy-related nutrients for bacterial colonization (El Asmar et al., 2000; Shi and Walker,2004; Hou et al., 2018). In addition, some important pathways related to Cellular processes,Environmental information processing, and Human disease functions also changed, indicating that BMD may aff ect the biochemical processes ofP.clarkiiintestinal bacteria at the cellular and molecular levels.
5 CONCLUSION
Overall, our study demonstrated that BMD causes changes in the intestinal f lora ofP.clarkii.Specif ically, we found that the prevalence of bacteria belonging to the phylum Proteobacteria in the intestine ofP.clarkiiincreased, while the prevalence of bacteria belonging to the phylum Bacteroidetes decreased, ref lecting the abnormal metabolism and nutrient absorption of crayf ish with BMD, which provides new insights for further research on the pathogenesis of BMD.
6 DATA AVAILABILITY STATEMENT
All raw data were submitted to the NCBI Sequence Read Archive (SRA) under accession number PRJNA692534. The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
 Journal of Oceanology and Limnology2022年5期
Journal of Oceanology and Limnology2022年5期
- Journal of Oceanology and Limnology的其它文章
- Comparison of three f locculants for heavy cyanobacterial bloom mitigation and subsequent environmental impact*
- Eff ect of light intensity on bound EPS characteristics of two Microcystis morphospecies: the role of bEPS in the proliferation of Microcystis*
- Community structure of aerobic anoxygenic phototrophic bacteria in algae- and macrophyte-dominated areas in Taihu Lake, China*
- Tidal water exchanges can shape the phytoplankton community structure and reduce the risk of harmful cyanobacterial blooms in a semi-closed lake*
- Eff ect of random phase error and baseline roll angle error on eddy identif ication by interferometric imaging altimeter*
- Estimating the evolution of sea state non-Gaussianity based on a phase-resolving model*