
Screening of nanobody against Microcystis from a human phage display nanobody library*
2022-10-08 02:18YaoZUWenjieMIAOYuLUOChenXUQianhuiPANSiyuCHENJianhongLI
Yao ZU, Wenjie MIAO, Yu LUO, Chen XU, Qianhui PAN, Siyu CHEN, Jianhong LI
School of Life Sciences, Nanjing Normal University, Nanjing 210046, China
Abstract Microcystis species identif ication is essential for ecological studies and water bloom control.Immunoassays are more specif ic and convenient and several approaches have been used to develop for diagnosing harmful red tide algae. However, investigations on Microcystis identif ication using immunological approaches are still in the initial stage. In this study, Microcystis aeruginosa PCC7806 lysates were utilized as coated antigens to enrich and screen specif ic Microcystis nanobodies from a human domain antibody display library. After three rounds of enrichment, 10 positive monoclonal particles were isolated from the library and the most two positive nanobodies (DAb2 and Dab3) were eff ectively produced in Escherichia coli BL21. Finally, the DAb2 showed specif ic immune binding to diff erent Microcystis by the immuno-dot blot assay. This antibody could be used to establish an immunological method to identify Microcystis.
Keyword: Microcystis; phage display library; nanobody; immuno-dot blot assay
1 INTRODUCTION
Harmful cyanobacterial blooms, in particularMicrocystisblooms, were the most pervasive in temperate and tropical freshwater bodies throughout the world (Xiao et al., 2018). Extensive species ofMicrocystiscan be responsible for harmful cyanobacterial blooms and lack eff ective analytical tools to identify them, making ecological monitoring research diffi cult. Until now, approximately 51 species ofMicrocystisgenus had been classif ied and listed in AlgaeBase (Guiry and Guiry, 2019), however, their identif ication remains controversial.
Traditional identif ication of microalgae relies on visible traits by microscope, including cell size,gel sheath form, cell arrangement in a colony, etc.(Komárek and Komárková, 2002). It is diffi cult to accurately identifyMicrocystisspecies only based on their morphologic characteristics because they vary depending on the environment, especially colonial appearance (Otsuka et al., 2000; Li et al., 2013; Sun et al., 2016; Xiao et al., 2018). Specif ic DNA region sequencing is a very useful technique for microalgal identif ication. However, this procedure is timeconsuming and cannot deliver results in real time.Additionally, when examining a water sample with mixed species under a microscope, the DNA analysis is ineff ective.
Immunological techniques based on specif ic antibodies are a rapid, sensitive, and simplif ied method for pesticides (Zhu et al., 2020), viruses(Bhardwaj et al., 2020), bacteria (Zhang et al., 2021),biotoxins (Szkola et al., 2014), and harmful algae detection (Blanco et al., 2015). Antibodies, both polyclonal and monoclonal antibodies, have been generated for varied algae, includingChlamydomonasreinhardtii(Mitra et al., 2012),Alexandriumminutum(Mendoza et al., 1995; Gas et al., 2009; Carrera et al., 2010), andPrymnesiumparvum(West et al.,2006). However, investigations onMicrocystisidentif ication using immunological approaches are still in the initial stage. Recently, Blanco et al. (2015,2017) have developed a validated and cost-eff ective 17-polyclonal antibody microarray (containing three antibodies againstM.aeruginosa,M.f los-aquae,andM.novacekii, respectively) for detecting and identifying cyanobacteria simultaneously. Only three polyclonal antibodies againstMicrocystiswere found to be insuffi cient in terms of the present variety ofMicrocystis.
In recent years, phage display library has become a promising technology for rapidly and effi ciently obtaining antibodies with high sensitivity and specif icity for food safety and environmental contamination detection (Zhao et al., 2016; García-García et al., 2020; Lee et al., 2020). The method works by randomly cloning artif icial antibody genes into phagemid vectors, then co-expressing them on the surface of phage capsid proteins, including single-domain antibody (sdAb), also known as nanobody (Nb) (Jiang et al., 2013; Qiu et al., 2018),single-chain antibody (scFv) (Xu et al., 2019a, b), and polypeptide (Lee et al., 2020). The candidate particles could be obtained by multiple rounds of biopanning from the library, and then their genes are sub-cloned into suitable vectors for over-expression (Xu et al.,2021). Nanobodies specif ic for surface antigens ofA.minutum,C.reinhardtii, andM.aeruginosawere isolated from pre-immune or immune phage display libraries and eff ectively employed in diagnostics(Jiang et al., 2013, 2014; Mazzega et al., 2019;Folorunsho et al., 2021).
In this study, we employed a large diversity and capacity of human domain antibody display library for screening specif icMicrocystisnanobodies. The most two positive nanobodies were expressed inEscherichiacoliBL21 and the DAb2 was specif ic for diff erentMicrocystis. The immuno-dot blot assay was used forMicrocystisdetection.
2 MATERIAL AND METHOD
2.1 Strain and reagent
MicrocystisaeruginosaPCC7806 andSynechocystissp. PCC6803 were obtained from Pasteur Culture Collection of Cyanobacteria, France.M.aeruginosaFACHB-905 was kindly provided by Dr. Han MENG (Nanjing Normal University).M.f losaquaeFACHB-1272,M.panniformisFACHB-1757,M.wesenbergiiFACHB-929,PediastrumduplexFACHB-1804 andChroococcussp. FACHB-193 were from Freshwater Algae Culture Collection at the Institute of Hydrobiology, Wuhan, China.MicrocystisXW01,Anabaenasp. JK12,Chlorellasp., andScenedesmussp. JK37 were isolated from blooms in Xuanwu Lake, Nanjing, China and Taihu Lake, China. All strains were cultured in liquid BG-11 medium (Waterbury, 2006) at 28±2 ℃ under continuous illumination at 1 500 lx.
Domain antibody (DAb) phage display library(based on a single human VH framework (V3-23/D47) with diversity introduced in the antigen-binding site and constructed in an ampicillin resistance pR2 phagemid vector, the size of the library is 3×109) was purchased from the Medical Research Council (MRC)Laboratory of Molecular Biology (Cambridge, UK)and used to pan specif icMicrocystisnanobodies.Helper phage M13K07 andEscherichia coliTG1 (E. coliTG1) were obtained from Nanobody(NB) Biolab (Chengdu, China).E. coliBL21 was purchased from Vazyme (Nanjing, China). Anti-HIS monoclonal antibody and His-Tag mAb (horseradish peroxidase (HRP) conjugated) were purchased from GenScript (Nanjing, China). Skim milk and trypsin were obtained from Sigma-Aldrich (Beijing, China).Six-well and 96-well plates were purchased from Corning (Beijing, China).
2.2 Preparation of protein extract
MicrocystisaeruginosaPCC7806 lysates were used as binding templates to select specif ic antibodies from the DAb phage display library. 50-mL of cells with an optical density of 0.6 at 720 nm (OD720) were harvested by centrifugation at 8 000×gfor 5 min at 4 ℃ and washed with phosphate-buff ered saline (PBS,pH 7.4) three times. The pellets were re-suspended in 1-mL PBS solution (pH 7.4) and frozen at -80 °C,then thawed the frozen repertoire on ice. Following triple freezing, the mixtures were centrifuged for 25 min at 8 000×gand 4 °C. The clear supernatant was collected and stored in small aliquots at -20 ℃until used. Protein concentration was estimated by the Bradford method (Bradford. 1976), using bovine serum albumin (BSA) as a standard.
2.3 Amplif ication of the DAb phage display library
Amplif ication of the antibody library was described with some modif ications in Xu et al. (2018). The frozen stock of phagemid inE.coliTG1 was thawed on ice and added into 250-mL 2×TY-AG medium(16-g tryptone, 10-g yeast extract, and 5-g NaCl in 1-L double distilled water with 100-μg/mL ampicillin and 1% glucose). The mixtures were cultured for 2 h at 37 ℃ and 250 r/min until cells density reached 0.6 at OD600, then KM13K07 helper phages (approximately 1012pfu/mL) were added and cultured for 1 h at 30 ℃in a water bath. The cultures were centrifuged at 3 200×gand 30 ℃ for 30 min, then precipitates were re-suspended with 500-mL 2×TY-AK (containing 100-μg/mL ampicillin and 50-μg/mL kanamycin) for growing overnight at 25 ℃ and 250 r/min. The next day, after being centrifuged for 30 min at 3 300×gand 4 ℃, a 500-mL supernatant was obtained, 125-mL PEG/NaCl (containing 20% polyethylene-glycol(PEG) and 2.5-mol/L NaCl) were added into the supernatant, then kept the mixtures on ice for 1 h. The mixtures were centrifuged for 30 min at 3 300×gand 4 ℃ and the precipitates were re-suspended with 5-mL sterile PBS solution and centrifuged at 11 600×gand 4 ℃ for 10 min. The supernatant was the amplif ied antibody library and the titer of phagemids was estimated by series dilutions of 10-fold and adjusted to 109CFU/mL for next study.
2.4 Enrichment and biopanning of phage particles binding to M. aeruginosa PCC7806 extract
Enrichment and biopanning of particles were performed as described with some modif ications by Xu et al. (2018). Six-well plate was coated with 2-mLM.aeruginosaPCC7806 lysates (the f irst round of screening was 200 μg/mL and the remaining two rounds were 100 μg/mL and 50 μg/mL) for standing overnight at 4 ℃. The next day, the plate was washed with 3-mL PBST solution (containing 0.1% Tween 20 in PBS solution) three times and blocked with 3-mL MPBS solution (containing 5% skim milk powder in PBS solution) for 2 h at 37 ℃. After washing three times with 3-mL PBST solution, 1-mL particles (2.13×109pfu/mL) mixed with 2-mL MPBS solution was added. The plate was shaken for 1 h at 120 r/min and room temperature, then standing for 1 h at 37 ℃. After being washed ten times with 3-mL PBST solution, unbound phages were removed and bounding ones were eluted by 1-mL trypsin solution(0.2-mg/mL trypsin in PBS solution). The eluent was the f irst round enriched library. Particles collected from every round were quantif ied at 109CFU/mL and evaluated by calculating the output/input and polyclonal phage enzyme-linked immunosorbent assay (ELISA) (Zhang et al., 2012).
According to the results of the output/input and polyclonal phages ELISA, 1.5-mLE.coliTG1 were infected by 0.5 mL of particles eluted in the third round, then spread on TYE-AG solid medium (containing 100-μg/mL ampicillin and 1%glucose) for culturing overnight at 37 ℃. The next day, individual colonies were randomly picked and inoculated into 96-well plates with 100 μL/well of 2×TY-AG medium for culturing overnight at 37 ℃. Two microliters of cultures were transferred to another plate for culturing 2 h at 250 r/min and 37 ℃, then 20 μL/well of KM13K07 helper phages(approximately 1012pfu/mL) were added into the plates for rescuing 2 h at 250 r/min and 37 ℃. The plates were centrifuged at 3 300×gfor 30 min at room temperature and the precipitates were re-suspended with 200-μL 2×TY-AK medium followed by incubating overnight at 250 r/min and 30 ℃. The next day, the plates were centrifuged at 3 300×gfor 30 min and the supernatants were used for monoclonal phage ELISA (Xu et al., 2018). A positive monoclonal phage antibody was considered according to the OD450ratio of P/N>3 (Wang et al., 2012). P/N=positive (coated with lysates) / negative (coated with carbonatebuff ered saline).
2.5 Colony PCR and DNA sequencing
The positive phage nanobody genes were identif ied by colony PCR and DNA sequencing. The PCR reaction conditions were as follows (primers were LMB3: CAGGAAACAGCTATGAC and pHEN:CTATGCGGCCCCATTCA): 98 ℃ for 3 min, 30 cycles of 98 ℃ for 10 s, 52 ℃ for 10 s and 72 ℃for 15 s, and f inal extension at 72 ℃ for 2 min. The amplif ied products were examined by 1% agarose gel electrophoresis and sequenced by Sangon Bio.Co. Ltd. (Shanghai, China). The alignment of the protein sequence was performed using DNAMAN 9.0 software.
2.6 Soluble expression and purif ication of nanobodies
The most two positive nanobody genes (DAb2 and DAb3) were cloned betweenNcoI andNotI sites of pET-29a(+) and pET-26b(+) vectors for soluble expression, respectively. In brief, the genes were amplif ied by PCR using primers DAb-NcoI-F CATGCCATGGGCCATGCCATGGGCCAGGT and DAb-NotI-R ATAAGAATGCGGCCGCGCTCGAGACGGTG, then the products were digested withNcoI andNotI and ligated into pET-29a(+) or pET-26b(+) vectors which had been digested with the same restriction endonucleases. The recombine plasmids were transferred intoE.coliBL21 and the positive clones were identif ied by colony PCR and DNA sequencing using universal primers (T7 promoter and T7 terminator). A well-sequenced single colony was inoculated into 5 mL of Luria-Bertani (LB) medium containing 50-μg/mL kanamycin and incubated at 37 ℃ overnight. Five milliliters of cultures were inoculated into 500 mL of LB medium containing 50-μg/mL kanamycin for culturing until cells density reached 0.6 at OD600, then adding isopropyl-β-Dthiogalactoside (IPTG) with a f inal concentration of 0.2 mmol/L to induce the antibodies expression for 16 h at 28 ℃ and 150 r/min. The cells were harvested by centrifugation at 8 000×gfor 10 min and 4 ℃ and re-suspended with 10-mL ice-cold lysis buff er (50-mmol/L sodium phosphate, 300-mmol/L sodium chloride, and 10-mmol/L imidazole, pH 8.0),then lysed by sonication on ice, working 15 s and intermittent 45 s for 15 min. After being centrifuged at 8 000×gfor 30 min and 4 ℃, the supernatant of whole cell lysate was used for preparing pure antibodies by a His60 Ni superf low resin & gravity column.The purif ied antibodies were dialyzed against PBS solution and concentrated to 3.5 mL using Amicon Ultra-15 centrifugal f ilter devices (Millipore). Then,the proteins were checked by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDSPAGE).
2.7 Gel electrophoresis and immunoblotting analysis
Prior to electrophoretic analysis, protein samples were boiled in SDS-PAGE buff er (300-mmol/L Tris-HCl, 10% w/v SDS, 30% v/v glycerol, 9.3% w/v dithiothreitol, and 0.012% bromophenol blue). Then,the proteins were separated with 10% SDS-PAGE gels and electrophoretically transferred to immobilon-P transfer membrane (ABM, catalog number B500).Prior to Western blot analysis, the membrane was blocked with 5% skim milk dissolved TBS solution with gentle rocking for 1.5 h at room temperature.The membrane was washed three times (15 min each)in TBST solution (containing 0.1% Tween 20 in TBS solution) and incubated with purif ied antibodies(DAb2 and DAb3) for 2 h at room temperature. After being washed with TBST solution, the membrane was incubated with Anti-HIS monoclonal antibody(1∶10 000 v/v dilutions in TBST solution) followed by a 1-h incubation at room temperature. After washing,the membrane was incubated with goat anti-mouse IgG (HRP) (1∶10 000 v/v dilutions in TBST solution)for 1 h at room temperature with gentle rocking. After intensive washing as noted above, immunoreactive proteins were visualized by incubation with enhanced chemiluminescent substrate (Tanon, catalog number 180-500) and images were generated by a cooled charge-coupled device (CCD) camera (Tanon-4100).
2.8 Protein extraction and immuno-dot blot analysis
About 107cells were broken with 50-μL PBS solution and 0.06-0.08-g glass beads (0.1-mm diameter)using a high-throughput tissue grinder (SCIENTZ)before being centrifuged at 10 000×gfor 10 min.Immuno-dot blot assay was performed to evaluate the nanobodies specif icity using the supernatant of diff erentMicrocystislysates (M.aeruginosaPCC7806,M.aeruginosaFACHB-905,MicrocystisXW01,M.f los-aquaeFACHB-1272,M.panniformisFACHB-1757, andM.wesenbergiiFACHB-929)and some companion species withMicrocystis(Synechocystissp. PCC6803,Chroococcussp.FACHB-193,Anabaenasp. JK12,Chlorellasp.,PediastrumduplexFACHB-1804, andScenedesmussp. JK37). In addition, a water sample collected from Taihu Lake with more than 90% ofMicrocystisbiomass was also used for detection. 5 μL of the supernatant were spotted onto immobilon-P transfer membrane (ABM, catalog number B500) and allowed to dry up at room temperature. After being blocked with 5% (w/v) skim milk in TBST solution for 1.5 h at room temperature, the membrane was washed three times (15 min each) and incubated with the nanobodies (5-μg/mL diluted in TBS solution) for 2 h at room temperature. After washing, the membrane was incubated with His-Tag mAb (HRP conjugated)(1∶5 000 dilutions in TBS solution) for 1 h at room temperature. After intensive washing as noted above, immunoreactive proteins were visualized by incubation with enhanced chemiluminescent substrate(Tanon, catalog number 180-500) and images were generated by a cooled CCD camera (Tanon-4100).
3 RESULT
3.1 Screening and identif ication of Microcystis particles
Comparing the third round enriched library to the original, the OD450value was increased approximately 6.3-fold by polyclonal phage ELISA (Fig.1). The result indicated that the target particles were enriched obviously. After three rounds of biopanning, about 960 clones (10×96-well plates) were analyzed by the phage ELISA and 10 positive particles whose OD450ratios of P/N>3 were obtained. The ratio of the most positive particle named DAb3 was 6.43 and the lowest named DAb5 was 3.90 (Fig.2a). All positive particles displayed a real and similar size (approximately 400 bp) gene fragments by colony PCR on a gel(Fig.2b). Then the particles were checked by DNA sequencing, four (DAb2, DAb3, DAb8, and DAb10)of which possessed diff erent amino acid sequences of the nanobodies displayed the same immunoglobulin frameworks from FR1 to FR4, but more or less diff erences in the complementarity determining regions from CDR1 to CDR3 (Fig.2c).
3.2 Nanobodies production and their immunoreactivity analysis
In this study, the most two positive antibodies(DAb2 and DAb3) were chosen and expressed inE.coliBL21, the expressed antibodies were abundant in the supernatant of whole cell lysate by ultrasonic breaking (Fig.3a). Consequently, the purif ied DAb2 and DAb3 antibodies were obtained from the supernatant of whole cell lysates by using a His60 Ni superf low resin & gravity column, which existed in a clear protein band (approximately 14.5 or 12 kDa) on SDS-PAGE gal as shown in Fig.3a. The concentration of the purif ied DAb2 and DAb3 in PBS solution were 506 and 972 μg/mL, i.e., 3.54 and 6.80 mg/L in the 500-mL original culture.
The immunoreactivity of purif ied antibodies was examined by Western blot. As shown in Fig.3b & c, both DAb2 and DAb3, exhibited positive immunoreactivity withM.aeruginosaPCC7806 polypeptides and they probably recognize the same antigen, only the diff erence in sensitivity modif ies the apparent distribution pattern.
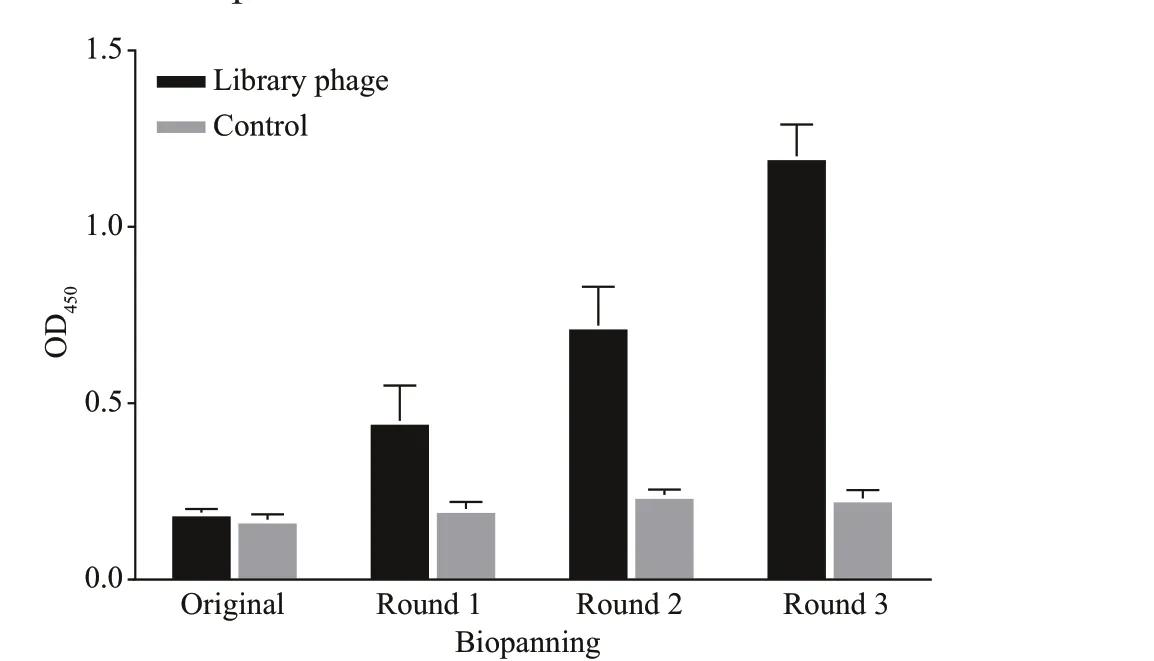
Fig.1 Polyclonal phage ELISA based on each round of the enriched library (quantif ied to 10 9-CFU/mL phage particles) for protein lysates of M. aeruginosa PCC7806
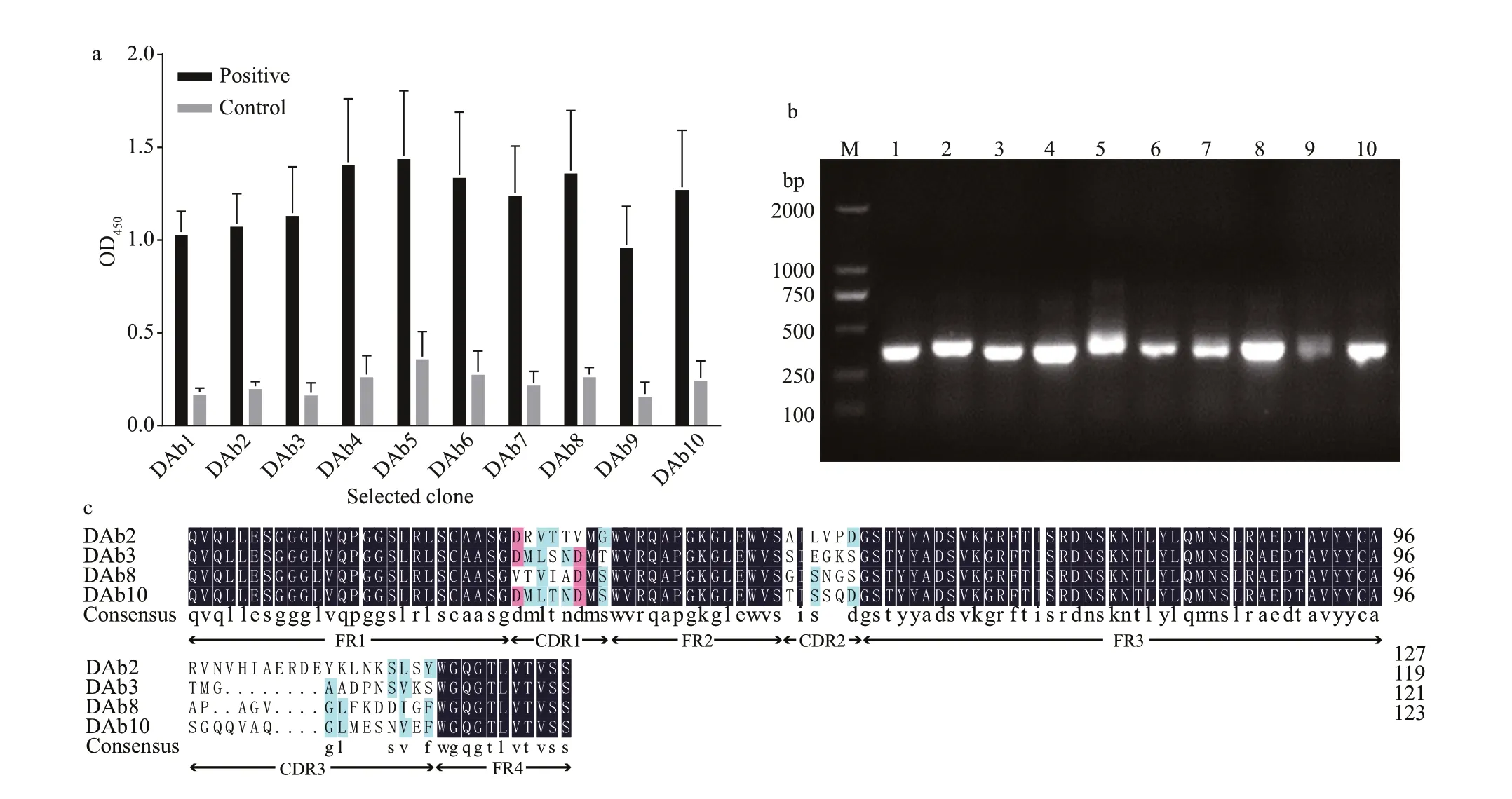
Fig.2 Monoclonal phage ELISA that the selected phage particles for protein lysates of M. aeruginosa PCC7806 (a); the colony PCR amplif ied products of the ten positive monoclonal phage antibody genes, which were named DAb1-10(b); amino acid sequences of the obtained positive monoclonal phage antibodies (c)
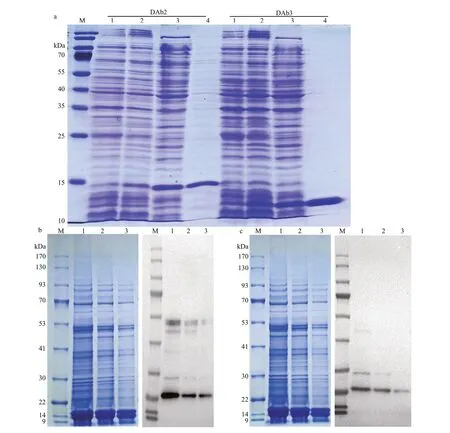
Fig.3 SDS-PAGE analysis of the two most positive nanobody proteins (DAb2 and DAb3) expressed in E. coli BL21 (a); the Western blot (WB) using purif ied nanobodies DAb2 (b) and DAb3 (c) as the detection antibodies
3.3 Antibodies specif icity and sensitivity of the immuno-dot blot assay
The DAb3 did not react with any algae examined in this study, therefore the DAb2 was used for all subsequent immunoassays. As shown in Fig.4a, the DAb2 bound effi ciently to all culturedMicrocystisstrains (M.aeruginosa,M.fl os-aquae,M.panniformis, andM.wesenbergii) and the f ield water sample. However, it exhibited an extremely weak signal forSynechocystissp. PCC6803 andChroococcussp., no cross-reaction with the other cyanobacterial and green algal strains (Anabaenasp. JK12,Chlorellasp.,PediastrumduplexFACHB-1804, andScenedesmussp. JK37). The detection limit of the immuno-dot blot assay was less than 240 cells ofM.aerginosaPCC7806(Fig.4b).
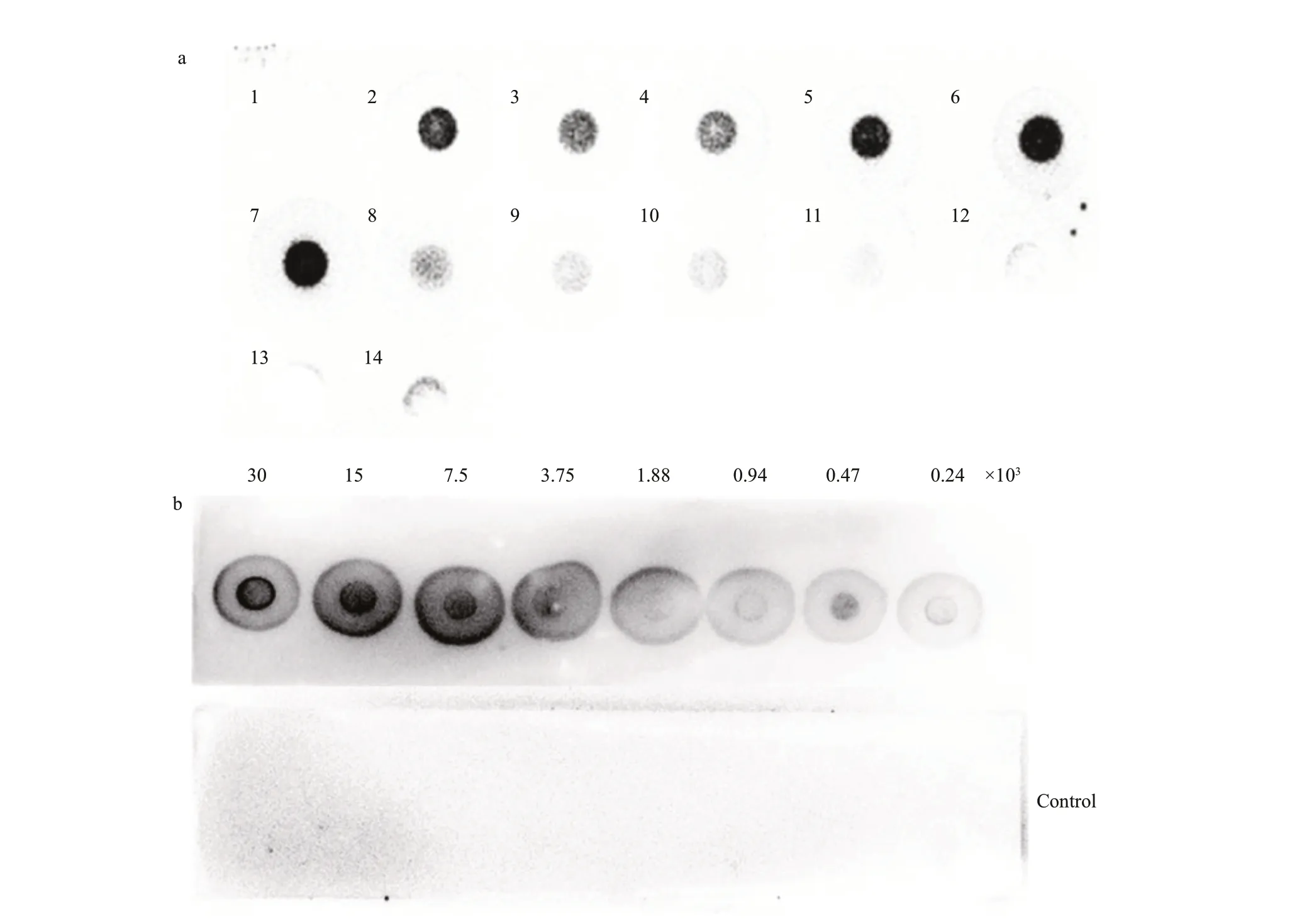
Fig.4 Various algae lysates were tested in the immuno-dot blot test to check the specif icity of antibody DAb2 (a); detection limit of the immuno-dot blot assay (b)
4 DISCUSSION
Microcystisidentif ication is mainly based on morphological characteristics determined by the microscopic method. It is a time-consuming and arduous process that requires specialist taxonomic knowledge. The immunological method could quickly and reliably identifyMicrocystisspecies and require only small sample volumes and a simple sample preparation based on a short lysis step or working with intact cells (Blanco et al., 2015, 2017;Folorunsho et al., 2021).
Polyclonal antibodies have been widely used.Blanco et al. (2015, 2017) have successfully produced three polyclonal antibodies againstM.aeruginosa,M.f los-aquae, andM.novacekii.However, the drawbacks of polyclonal antibody including burdensome immune programs and diffi culty in reproducing should be unavoidable.Additionally, when animals injected with toxic cyanobacterial cells or lysates, the target antibodies could not be got due to the toxicity of antigen (Gas et al., 2009). Phage display antibody library has become a promising technology to rapidly obtain specif ic antibodies for any antigen, independently of its toxicity or immunogenicity by a fast in vitro high-throughput screening without immunization(Xu et al., 2018). In this study, we selected specif ic nanobodies againstMicrocystisby panning in vitro a human domain antibody display library and then conveniently converted them into applicable reagents for diagnostics. Our goal is to mass-produce a genetic engineering antibody, which could provide a guarantee for the development of immune methods forMicrocystisdetection.
The species selectivity of the anti-Microcystisnanobody (the DAb2) produced in this work enabled to use it for the detection ofMicrocystiscultivated in the laboratory and grown in the f ield. Although the DAb2 could recognize allMicrocystistested in this study, it should be emphasized the affi nity of the DAb2 varied among the diff erentMicrocystisspecies. It exhibited higher immunoreactivity withM.aeruginosastrains (PCC7806 and FACHB-905),M.f los-aquaeFACHB-1272,M.panniformisFACHB-1757,andMicrocystisXW01 thanM.wesenbergiiFACHB-929 (Fig.4a). Previous studies showed thatM.wesenbergiican be distinguished from otherMicrocystisbecause of the signif icant gel sheath of colonies and divergence in thecpcBA-IGS sequence(Komárek and Komárková, 2002; Tan et al., 2010).The results indicated thatM.wesenbergiicould have a distant evolutionary relationship with otherMicrocystisspecies. The DAb2 weakly recognizedSynechocystissp. PCC6803 andChroococcussp.,both of which belong to the same Chroococcales asMicrocystis. The faint signal could be exploited as a negative control in the detection ofMicrocystisby the immuno-dot blot assay. Recently, Folorunsho et al. (2021) have selected three specif ic nanobodies againstM.aeruginosaby whole-cell biopanning from a naïve phage display library. However, whether the antibodies reacted with the otherMicrocystisspecies were not clear. The DAb2 could be more eff ectively applied for simultaneous detection ofMicrocystisspecies.
The immuno-dot blot assay based on the DAb2 showed that 240 cells ofM.aeruginosaPCC7806 displayed an obvious signal, it is inferred from the signal that the detection limit could be less than the 200 cells, similar to Blanco’s polyclonal antibody(the detection limit was about 102-103cells) (Blanco et al., 2015). However, it existed a big gap compared with the records by Folorunsho et al. (2021) that reached to 1.1 and 2.2 cells/mL for the strainsM.aeruginosaPCC7806 and PCC7005. To improve the detection sensitivity, some signal amplif ication strategies would be introduced, such as using antibody-conjugated AuNPs (Thiruppathiraja et al.,2011) and f luorescent labeled-antibody (Yu et al.,2011; Niu et al., 2012). In addition, more sensitive detection methods, such as f low cytometry based on f luobodies (Mazzega et al., 2019), thermal lens spectrometry (TLS) diagnostic method (Folorunsho et al., 2021), and electrochemical immunodetection(Adkins et al., 2017) could be used in the immune detection ofMicrocystisspecies.
5 CONCLUSION
In this study, we have screened a specif ic nanobody DAb2 againstMicrocystisfrom the human domain antibody display library. The DAb2 intensively recognizedM.aeruginosa,M.f los-aquaeandM.panniformis, and weakly recognizedM.wesenbergiiby the immuno-dot blot assay. The antibody could be used forMicrocystisimmunodetection.
6 DATA AVAILABILITY STATEMENT
The data generated in this study are available from the corresponding author or the f irst author upon reasonable request.
 Journal of Oceanology and Limnology2022年5期
Journal of Oceanology and Limnology2022年5期
- Journal of Oceanology and Limnology的其它文章
- Comparison of three f locculants for heavy cyanobacterial bloom mitigation and subsequent environmental impact*
- Eff ect of light intensity on bound EPS characteristics of two Microcystis morphospecies: the role of bEPS in the proliferation of Microcystis*
- Community structure of aerobic anoxygenic phototrophic bacteria in algae- and macrophyte-dominated areas in Taihu Lake, China*
- Tidal water exchanges can shape the phytoplankton community structure and reduce the risk of harmful cyanobacterial blooms in a semi-closed lake*
- Eff ect of random phase error and baseline roll angle error on eddy identif ication by interferometric imaging altimeter*
- Estimating the evolution of sea state non-Gaussianity based on a phase-resolving model*