
癌症多组学数据深度自编码器整合分型方法
2022-09-21 05:37曹业伟
计算机工程与应用 2022年18期
曹业伟,刘 飞
华南理工大学 软件学院,广州510006
癌症又称为恶性肿瘤,是一系列由于细胞不受控制地生长分裂导致的疾病。癌症往往由基因突变引起,并伴随着细胞层面的分子改变[1],包括基因表达、DNA 变异、RNA表达改变等。因此,随着高通量测序技术的发展,在分子水平上对癌症重新分类和分型已逐渐变得至关重要。但是在细胞层面上,单一组学数据能提供的信息较为有限。为了有效解决这个问题,研究人员开始对海量的生物多组学数据(例如基因组学数据、转录组学数据、表观遗传学数据、蛋白质组学数据等)[2]进行研究和分析。利用多组学数据对癌症亚型进行有效判别,可以在评估患者的癌症转移情况和选择治疗手段方面起到重要作用[3],促进精准医学的发展。
目前,癌症多组学数据的有关研究已取得许多重要突破,出现了各类生物学数据库,例如癌症体细胞突变目录(the catalogue of somatic mutations in cancer,COSMIC)数据库[4]、国际癌症基因组协会(international cancer genome consortium,ICGC)[5]、癌症基因组图谱(the cancer genome atlas,TCGA)[6]和儿童癌症有效疗法研究与应用(therapeutically applicable research to generate effective treatments,TARGET)[7]等,这些数据库为癌症研究分析提供了大数据基础。通过高通量测序技术和多组学数据整合方法,将基因组学、转录组学、表观遗传学和其他各类组学数据与癌症发生发展相结合起来,可以在分子层面探索癌症的发病机理。与以往的单组学数据相比,多组学数据整合可以利用各种数据对癌症信息交叉补充,不仅可以更好解决单组学信息缺失问题,也有助于研究人员从更多维度研究癌症。
目前针对整合多组学数据有一系列的研究。传统的多组学整合大多采用统计学方法。例如基于隐变量进行整合,有学者提出了iClusterPlus[8]方法,针对不同数据类型(二值型、离散型和连续型)分别建立隐变量与观测值的正则化的回归模型,接着基于隐变量对样本进行聚类分析,是一种隐变量求解模型,这类方法最大的问题是一旦多组学数据之间的维度差异过大或者样本数量较少时会难以拟合隐变量,同时也十分依赖特征选择,需要较多先验知识选择多组学特征。除了借助隐变量,还有采用相似度网络进行整合的方法,例如相似性网络融合(similarity network fusion,SNF)[9]和高阶路径相似度网络的融合模型(high-order path elucidated similarity,HOPES)[3]通过构建不同样本之间的相似度网络,并不断迭代更新相似度网络来整合多组学数据,这一类算法的优点是引入了相似度网络,使得不同组学数据的维度差异更加均衡,改善了隐变量模型的缺陷。最近在生物信息学中引入的深度学习技术也被用于多组学数据整合任务。Kim 等人[10]设计了一个进化神经网络来评估卵巢癌的预后情况。Chaudhary等人[11]通过自编码器(Autoencoder)整合了RNA、miRNA和DNA甲基化数据,可以从肝癌数据中提取压缩特征,采用Cox 模型根据生存时间对压缩特征进行筛选,得到具有新特征的样本。对样本进行聚类得到标签,利用标签监督训练SVM分类模型。相同的整合流程也被用于神经母细胞瘤[12]和头颈部鳞状细胞癌[13],并成功识别出了两种不同预后效果癌症亚型。Chai 等人[14]使用该方法对12 种癌症数据集测试,均可以识别出高风险和低风险患者组。对于具有高维度、高噪声的多组学癌症数据集,由于深度神经网络有较强的拟合能力,所以这些基于深度学习的多组学整合方法相比传统方法更具优势。但是这一套多组学整合流程仅识别出两种癌症预后亚型,即高风险组和预后低风险组,并且还要利用生存时间来选择特征,这意味着如果缺乏临床生存数据则难以进行研究,本质上还是一种监督学习模式。Jonathan 等人使用变分自编码器(variational autoencoder,VAE)对结肠腺癌进行癌症分型,并得到5种分子亚型[15],但分型结果与已有的生理学分型并不太吻合,缺乏生物学支撑,效果较为一般。
在本研究中一共采用了三种多组学数据,分别是基因表达、DNA甲基化和miRNA表达。其中基因表达是一类重要的生物学数据,基因经过转录表达生成相应产物,这些产物进行各类生化反应;DNA甲基化是一种比较有代表性表观遗传学数据,甲基化虽然不改变基因本身,但是会改变被修饰基因的表达情况,一般可以作为公认的癌症标志;miRNA 主要通过与mRNA 结合进而调控基因的转录情况,也是一种表观遗传学数据。这三种组学数据可以互相补充生物学信息,从而可以更全面地了解患者的状态。针对这三类癌症多组学数据,本文提出了一种基于深度自编码器的多组学整合方法(deep learning for multi-omics integration,DAEMI)。该方法的主要思想为:将多组学数据输入自编码器中进行重构训练,然后从瓶颈层中获取新的特征,对于具有新特征的样本,使用K均值算法对其聚类,进而识别癌症亚型。在模拟数据集与真实癌症数据集上分别测试了该方法,并与其他四种方法:高阶路径相似度网络的融合模型(HOPES)[3]、相似性网络融合(SNF)[10]、iClusterPlus[8]和moCluster[16]进行了比较。结果表明DAEMI聚类效果优秀稳定,抗噪声能力强,并可以取得具有显著生存差异的癌症亚型分组,同时比其他方法表现得更好。
1 多组学整合研究方法
1.1 实验数据介绍
本文采用了两类数据集进行相关整合算法的测试,分别是模拟数据集和真实癌症数据集。模拟数据集的设置目的是在具有人工标注标签的情况下,精确评估本文算法的聚类效果。而真实癌症数据集是为了进一步验证本文算法的在真实场景下的应用能力,能否应用于生物工程的研究、癌症预后研究等实际科研领域。
1.1.1 模拟数据集介绍
模拟数据集使用真实组学数据与预设聚类结构相结合的方式,构建方式与先前的一项研究[3]类似。其中真实数据来自GEO,选取了DNA 甲基化、基因表达和miRNA表达,GEO代码分别为GSE51557、GSE73002和GSE106453。为了尽可能地模拟真实生物数据的维度差异和噪音水平,通过SVD 分解将真实数据与预设聚类结构融合得到相应模拟数据集。构造的模拟数据集设定为无法通过单一组学得到相应真实分组,必须对三种组学进行整合才可以得到真实标签。为了进一步加大聚类难度,使用均值为0可变方差的高斯噪声对模拟数据集进行污染,模拟复杂多组学数据的背景噪音,根据方差大小分为低噪音、中噪音和高噪音三个级别。
1.1.2 真实癌症数据集介绍
在本研究中,除了模拟数据集,还使用了来自癌症基因组图谱(TCGA)的真实癌症数据集。一共有四类癌症,分别是结肠腺癌(colon adenocarcinoma,COAD)、多形胶质母细胞瘤(glioblastoma multiforme,GBM)、肾透明细胞癌(kidney renal clear cell carcinoma,KIRC)和肺鳞状细胞癌(lung squamous cell carcinoma,LUSC)。每个真实数据集都包含三种类型的组学数据:DNA 甲基化、基因表达和miRNA 表达。这四个数据集曾被用于高阶路径相似度网络的融合模型(HOPES)[3]和相似度网络融合(SNF)[10]研究。
1.2 多组学数据整合流程
DAEMI 的工作流程如图1 所示。受到文献[17]启发,借助深度自编码器提取特征能力,本文中使用深度自编码器进行重构训练,学习多组学数据的特征表示。在此工作流程中,将DNA 甲基化、基因表达和miRNA表达数据输入到具有3个隐藏层的深度自编码器中,然后从瓶颈层提取所需的压缩特征,使用K均值算法对所有这些具有新特征的样本进行聚类,从而得到不同的癌症亚型。
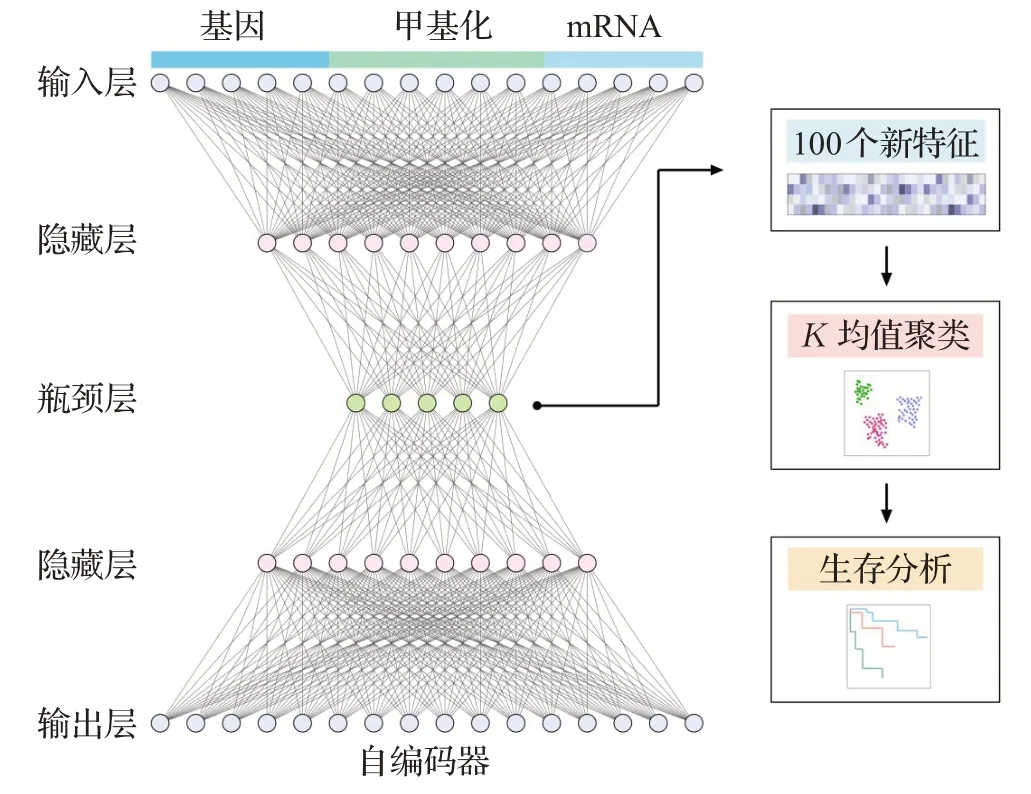
图1 多组学整合的流程结构Fig.1 Architecture of multi-omics integration
与以前的研究相比[11],本文中的方法省去了使用生存数据选择特征的步骤,从而可以避免了对生存数据的依赖。不同于Chaudhary 等人[11]的做法,没有使用Cox模型筛选特征,而是认为所有新特征均与癌症的预后亚型分型有关,因此可以扩大使用特征的范围,更好地利用所有信息。相比自编码器输出层的重构数据,更需要自编码器从输入重构过程中学习到的特征表示,即隐藏层的输出。因为多组学输入信息维度过高,需要较小的隐藏层进行降维,这样迫使自编码器学习到相关多组学特征。自编码器的重构输出由于仍然是高维度信息,实用价值不大,故没有在实验中使用。
1.3 深度学习框架
自编码器是一种无监督的前馈非递归神经网络,自编码器由输入层、隐藏层和输出层组成[18],具有多个隐藏层的自编码器即为深度自编码器。给定一个样本x=(x1,x2,…,xn),每个样本具有n维特征,则自编码器的目标是将x重构为x′。使用sigmoid 函数作为激活函数:
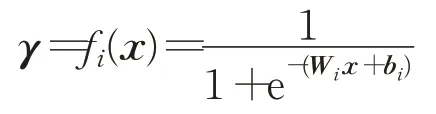
其中,x和γ分别是大小为d和p的两个向量。Wi是权重矩阵,bi是偏置矩阵。自编码器根据以下公式计算x′:
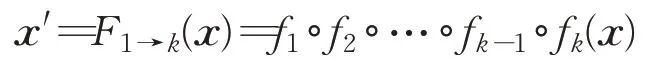
其中,k是网络层数,在本文中设置为3 层。相邻两层fk-1与fk的计算过程为一个组合函数fk-1∘fk(x)=fk-1(fk(x))。选择均方误差作为损失函数:
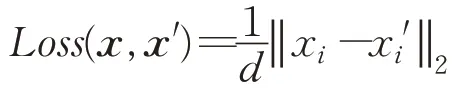
在本文实验设计环节,采用Python 语言编写程序,利用深度学习框架Keras实现自编码器[19]。选择了具有3个隐藏层的深度自编码器,将瓶颈层中学习到的特征作为样本的新特征。对于模型的优化求解过程采用了自适应矩估计算法[20]计算每个参数的自适应学习率。
1.4 K 均值聚类
癌症多组学数据整合工作完成后,需要开展相应的下游聚类任务,本文中采用的聚类方法为K均值聚类。给定样本集D=(F1,F2,…,Fn),K均值聚类方法将数据划分为K类(C1,C2,…,CK)。给定聚类簇数K时,簇CK代表簇中nk个对象的集合[21]。第j个簇Cj的质心为:
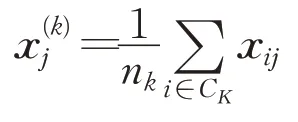
癌症数据集样本经过自编码器重构训练后,将瓶颈层中得到的压缩特征替换为样本的新特征,再通过K均值算法对这些具有新特征的样本进行聚类,得到相应的癌症分型结果。为了更好地与之前的算法(例如SNF和HOPES)进行比较,本文设置了相同数量的聚类簇数,即3个,代表可以识别3种癌症预后亚型。本文中的K均值算法通过Python第三方软件包scikit-learn[22]实现。
1.5 生存分析
受限于真实癌症数据集是无标签数据,针对聚类得到的癌症分型结果,参考先前多组学整合研究[3,11-12],大多利用生存分析来验证癌症分型结果,本文中使用了Kaplan-Meier生存估计[23]和时序检验[24]两种生存分析方法,评估聚类得到不同组之间是否有显著的生存差异。Kaplan-Meier方法通过估计生存函数,可以绘制一组病例的生存曲线,不同组别的生存曲线分隔越大,生存曲线交叉越少,则代表生存差异越明显。将聚类得到的三种组别的Kaplan-Meier曲线绘制出来,并计算了每种癌的时序检验P值。生存分析采用Python第三方软件包lifelines实现[25]。
2 实验分析
2.1 实验数据与预处理
为了验证深度自编码器的多组学整合能力,分别采用模拟数据集和真实数据集进行测试。
模拟数据集通过SVD 分解构建而成,模拟数据集预设四种聚类结构。其参数如下:样本数量为200 个,样本特征为3 980个。选取均值为0,标准差为2.4、2.7、3的高斯噪声对数据集进行污染,分别设定为低噪音数据集、中噪音数据集、高噪音数据集。
针对真实数据集,本文选择了缺失值不超过20%的样本,对于其他缺失较少的病例样本使用插值法来填充缺失值。四类癌症数据集的主要参数如下:结肠腺癌(COAD)数据集有92 个有效样本,41 214 个特征;多形胶质母细胞瘤(GBM)数据集有215个有效样本,13 881个特征;肾透明细胞癌(KIRC)数据集有122 个有效样本,43 188 个特征;肺鳞状细胞癌(LUSC)数据集共有106个有效样本,35 468个特征。
本文对实验数据均采用z-score标准化,以减弱不同组学间的差异。将经过标准化的数据输入到深度自编码器中进行重构训练。每一个样本将三种组学特征直接拼接而成,例如DNA 甲基化有1 000 维特征,基因表达有500维特征,miRNA有1 500维特征,那么最终输入自编码器模型的样本有3 000维特征。
2.2 模拟数据实验
在模拟癌症数据集实验中,将隐藏层中的节点数分别设置为500、100、500,新的压缩特征从具有100 个节点的瓶颈层中提取得到。训练次数为50轮。
模拟实验结果采用归一化互信息(NMI)进行评价,NMI可以衡量两个聚类结果的相近程度,结果越接近1,相近程度越高。汇总实验结果如表1 所示。与传统方法相比,可以看出本文的方法具有最好的聚类效果,在各类噪声情况下表现优异,抗噪声能力很强。总体综合来看,聚类结果领先SNF、iClusterPlus 和moCluster 很多,与HOPES 相比在低噪声、中噪声数据集领先,在高噪声数据集表现更加优秀。其中SNF 随着噪音强度提升,NMI 下降明显。iClusterPlus 和moCluster 虽然抗噪声能力很强,但是整体NMI处于较低水平,整合效果一般。HOPES虽然效果好,但是随着噪声增加,下降幅度比较大,鲁棒性一般。而本方法对噪声有很强的抵抗能力,同时整体表现较好。
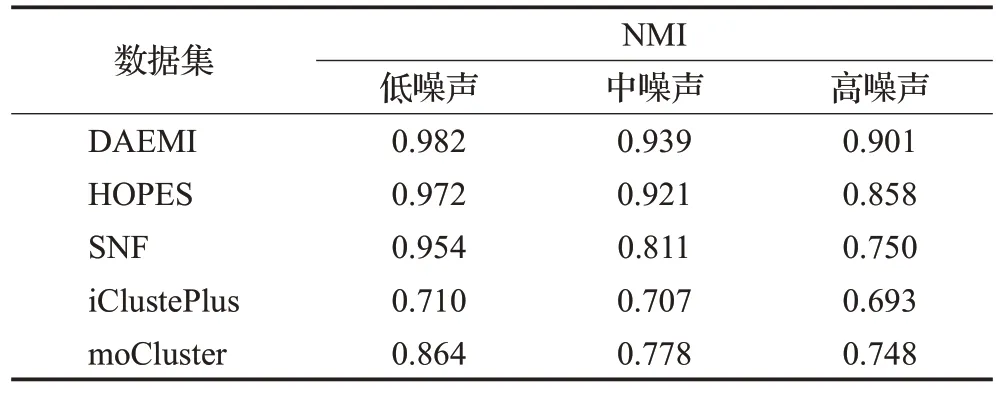
表1 DAEMI方法与HOPES、SNF、iClusterPlus和moCluster的模拟数据集表现对比Table 1 Performances on simulation dataset of DAEMI compare with other approaches:HOPES,SNF,iClusterPlus and moCluster
总体来说,本文方法可以在模拟数据集上取得很好的结果,能有效证明对于多组学数据的挖掘整合能力。
2.3 真实癌症数据实验
在真实癌症数据集实验中,隐藏层节点数目与模拟实验设置一致。训练次数为100 轮。受限于真实癌症数据缺乏真实标签,参考各类多组学整合研究的主流评价方案[3,11-12],借助临床数据绘制出生存曲线,并计算相关时序检验值,时序检验值可以判断多组生存曲线之间是否有显著差异,汇总时序检验值如表2所示。结合生存曲线与时序分析值作为癌症预后亚型分型评估标准。不同组别的生存曲线间隔越明显,证明生存差异越大。
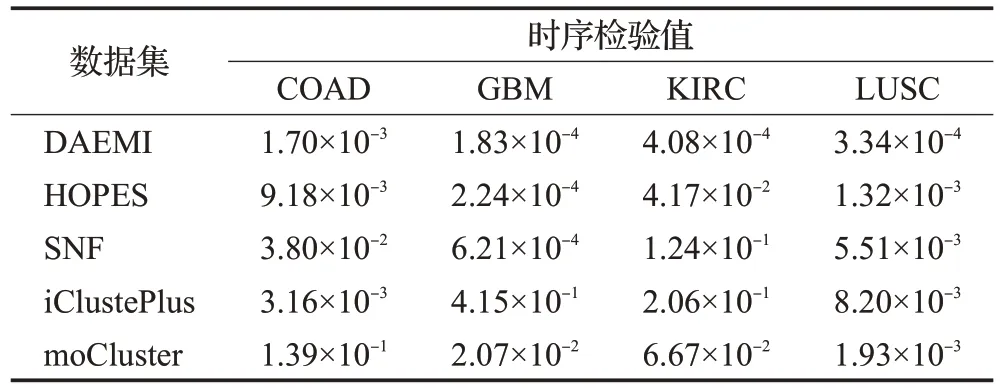
表2 DAEMI方法与HOPES,SNF,iClusterPlus和moCluster的真实癌症数据集表现对比Table 1 Performances on cancer datasets of DAEMI compare with other approaches:HOPES,SNF,iClusterPlus and moCluster
对于每一种癌症,通过聚类得到三种不同的癌症预后亚型,并绘制了每种亚型的Kaplan-Meier 生存曲线,如图2 所示。根据生存数据计算了每种癌症的时序分析P 值,并与先前的研究进行了比较,结果如表1 所示。图2的Kaplan-Meier曲线显示每种癌症亚型之间均存在显著生存差异(时序分析P值<0.05),查看生存曲线图,三条癌症亚型的Kaplan-Meier生存曲线之间分隔明显,交叉情况较少,可以看出确实存在着生存上的差异性。横向对比四种癌症数据集的结果,结合生存分析曲线和时序分析值来看,DAEMI在LUSC数据集上效果最好,其时序分析P值为3.34×10-4。
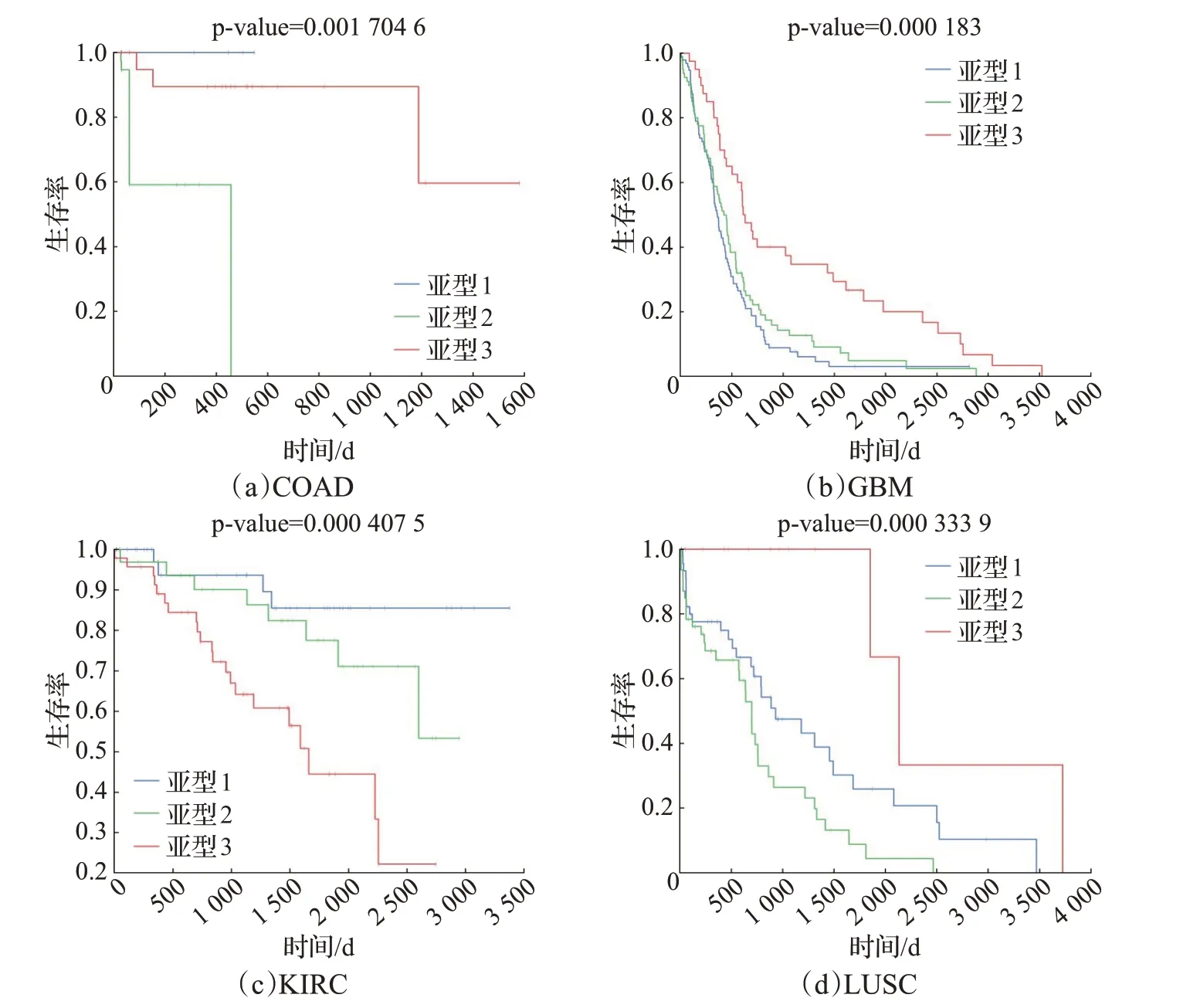
图2 TCGA中4种癌症的生存曲线Fig.2 Survival curves for 4 cancers from TCGA
与各类整合方法对比,通过生存分析检验癌症不同亚型之间是否存在显著生存差异。将本文的方法与先前研究的传统方法进行比较,由于前文提及的深度学习整合方法是需要生存信息进行特征筛选,之后再进行SVM监督学习训练分类,本质上是一种监督学习方式,所以无法参与本次实验比较。
生存曲线图如图2 所示,直观来看DAEMI 整合的癌症数据集的三条生存曲线分隔比较明显,交叉现象较少,没有出现生存曲线交叉缠绕的现象,这证明三种亚型之间的预后确实存在着明显区别。汇总五种方法时序分析结果如表2 所示,可以看到DAEMI 均具有最好的时序分析P值,时序分析值与相应的生存曲线图结合判断,验证了DAEMI 得到的三种亚型之间确实存在着显著的生存差异。
值得指出的是,仅有GBM 数据集,DAEMI 略好于其他方法,而在COAD、KIRC 和LUSC 三个数据集上,本文的结果均明显优于其他方法。这是由于COAD(维度41 214)、KIRC(维度43 188)和LUSC(维度35 468)数据维度远大于GBM数据集(维度13 881),并且GBM数据集样本数目最多(215 个),所以COAD、KIRC 和LUSC 的聚类难度远大于GBM 数据集,这也印证了相应的实验结果,表明DAEMI对高维度、少样本的真实数据集的效果很好,具有很好的实用性。DAEMI在KIRC数据集上优势明显,这是聚类难度最大的数据集,其余方法均产生了较大的时序分析值,这表明DAEMI 发现癌症分子亚型的表现很好,对于各类癌症都具有很好的泛用性。尽管HOPES、SNF、iClusterPlus和moCluster方法在某些癌症(例如LUSC 和GBM 数据集)中表现良好,但是缺乏一定的泛用性,例如在KIRC数据集上表现很差,表现不够稳定,这是由于iClusterPlus 方法十分依赖特征筛选,需要配合相应的先验知识,同样的问题也出现在moCluster 上。HOPES 和SNF 方法相比前两者有一定提升,但效果仍一般,泛用性比较一般。总的来说,本文方法在不同种类的癌症数据集上表现更可靠,在聚类有效性和聚类稳定性方面都优于现有方法。
2.4 功能分析
2.4.1 393种差异表达基因
以KIRC 数据集为例进行功能分析,并使用R 包EBseq[26]对三组患者进行差异基因表达分析。
EBseq可以对多个组进行差异基因表达分析,找出在不同癌症预后亚型中有明显表达差异的基因。在本研究中,由于K均值聚类中获得了3 个癌症亚型分组,所以假设基因表达中有3 种不同的状态,分别是低表达、中表达和高表达。因此,如图3所示有5种可能的表达模式(P1、P2、P3、P4、P5)。其中,模式P1 表示三个组中基因表达状态均一致;模式P2 表示三个组中前两组基因表达状态一致,第三组表达情况与前两组不同;模式P3 表示第一组和第三组表达状态一致,第二组表达情况不同;模式P4、模式P5以此类推。从中找出3个组中表达状态都不相同的基因,即处于模式P5 的基因。经过EBseq 处理,可以发现393 个差异表达的基因处于模式5,其后验概率均大于0.9,因此把这393 个基因作为差异表达的基因。
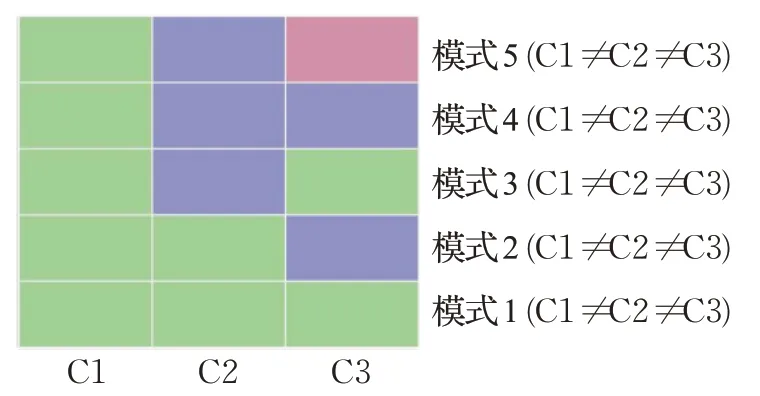
图3 五种表达模式Fig.3 All 5 possible expression patterns
图4绘制了393个差异表达基因的热力图。选取了相关的具有代表性的差异表达基因,例如UQCRC1、AP1M2、RAB25、HIGD1A,这些基因在先前的研究中已有报道。UQCRC1 是透明细胞肾细胞癌的生物标志物[27],AP1M2 也与透明细胞肾细胞癌有关[28],RAB25 是在胃肠道粘膜,肾脏和肺中表达的小GTP 结合蛋白[29],HIGD1A 曾在大肠癌研究中报道[30]。从中可以看到深度多组学整合方法可以有效地进行多组学整合和癌症分型任务,同时也具有相关癌症研究的生理意义。
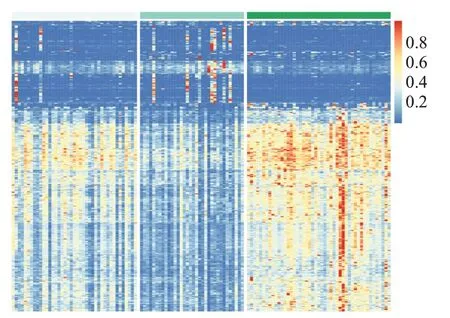
图4 393种差异表达基因热力图Fig.4 Heat map of differentially expressed 393 genes
2.4.2 富集通路分析
对于这393 个基因,使用了R 包clusterProfiler[31]进行了KEGG[32]通路分析和GO[33]通路分析,绘制出富集通路气泡图。其中纵坐标是各类通路;横坐标为比率,代表该通路下差异基因占差异基因总数的比例。右侧图例中,气泡大小代表基因个数,气泡越大,基因个数越多;气泡颜色代表富集的显著程度,颜色越红,显著程度越高。图5显示了11个富集的GO项(Benjamini-Hochberg P 值<0.05)。横轴表示富集倍数(Fold.Enrichment),纵轴从上到下依次是线粒体内膜(mitochondrial inner membrane)呼吸链(respiratory chain)、线粒体ATP 合成耦合电子输运(mitochondrial ATP synthesis coupled electron transport)、ATP合成偶联电子传输(ATP synthesis coupled electron transport)、线粒体呼吸链(mitochondrial respiratory chain)、呼吸链复合体(respiratory chain complex)、氧化还原酶复合物(oxidoreductase complex)、线粒体呼吸链复合体(mitochondrial respiratory chain complex I)、NADH 脱氢酶复合物(NADH dehydrogenase complex)、呼吸链复合体(respiratory chain complex I)、液泡质子转运V 型ATP 酶复合物(vacuolar proton-transporting V-type ATPase complex)。这些通路中,如线粒体内膜、线粒体ATP合成、电子传递和线粒体呼吸链等均与线粒体有关。参考先前的研究,线粒体在癌症的发展和转移中起着重要作用[34],可以看出DAEMI整合方法能有效定位相关基因通路。
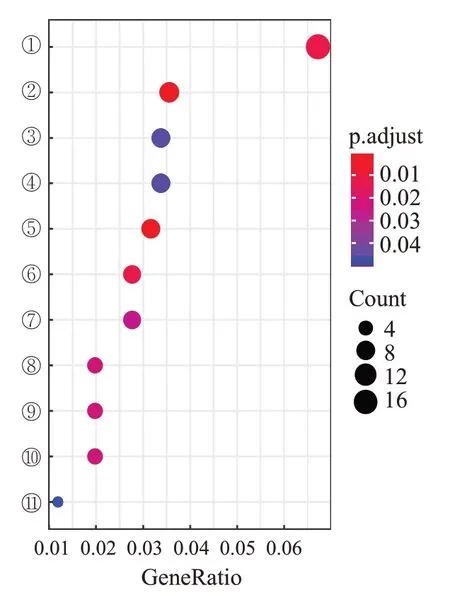
图5 GO富集通路Fig.5 GO enriched pathways
如图6 所示,在KEGG 分析中确定了8 种通路(Benjamini-Hochberg P值<0.05),纵轴从上到下依次是氧化磷酸化(oxidative phosphorylation)、亨廷顿病(huntington disease)、帕金森综合症(Parkinson disease)、非酒精性脂肪肝病(non-alcoholic fatty liver disease,NAFLD)、老年痴呆症(Alzheimer disease)、心肌收缩(cardiac muscle contraction)、收集导管酸分泌(collecting duct acid secretion)、同源重组(homologous recombination)。其中最重要的通路是氧化磷酸化,这已被先前的研究证实与癌症发展有关[35]。KEGG 富集分析还揭示了一系列神经退行性疾病通路与癌症发展有关,如亨廷顿病、帕金森病和阿尔茨海默氏病。先前的研究[36]表明神经退行性疾病可能与癌症共享某些通路,例如线粒体功能障碍和氧化应激在神经退行性疾病和癌症中均起关键作用[37]。
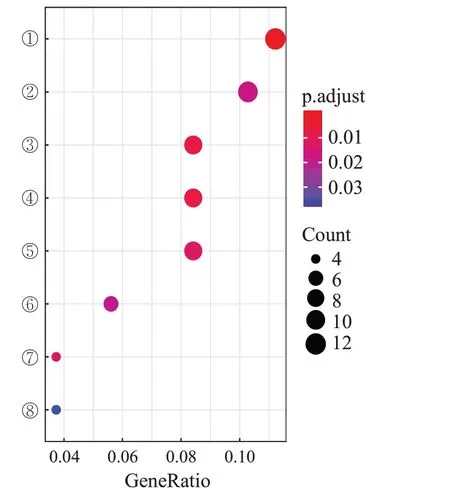
图6 KEGG富集通路Fig.6 KEGG enriched pathways
3 结束语
随着高通量技术的发展,多组学数据整合方法的探索可以促进癌症亚型的研究。先前研究提出的传统方法易受到噪声的影响,因此在某些癌症数据集上的效果较差,不具备一定的普适性和鲁棒性。而目前多数基于深度学习的多组学整合方法,依赖生存数据进行特征筛选,仅能对两种癌症亚型的识别效果较好,对多种癌症亚型的识别效果较差。
为了解决各种癌症亚型的分类问题,提出了一种基于深度自编码器的多组学数据整合方法,即DAEMI。使用自编码器整合DNA甲基化,基因表达和miRNA表达,从瓶颈层提取新特征,再进行聚类分析得到三种亚型。与其他传统研究方法对比[3,8,10,16],本文的方法在模拟数据集与真实癌症数据集上均取得了优良的结果,在数据量少的癌症数据集上优势更加显著。这表明DAEMI比传统方法更加有效,能够适应不同的癌症数据集,在多种癌症数据集上表现结果比较接近,具有很好的泛用性。与依靠生存时间来选择特征的深度学习方法相比[11-14],本文的方法不需要筛选特征,并且可以识别更多的癌症亚型。DAEMI使用了深度自编码器来提取新特征,不需要特征筛选就可以完成癌症分型任务,在缺失临床数据的数据集上也能够进行亚型分析。
目前本文的方法还需要在更多癌症数据集上进行测试,同时也需要进行更多不同组学数据的对比测试。多组学整合的流程一般是进行数据整合,再开展下游的聚类任务。本研究是将自编码器应用在整合部分,相当于对多组学数据进行特征提取。目前正在做的工作是进一步将整合任务与聚类任务进行联合优化,利用深度聚类方法同时完成数据整合和聚类标签输出两个任务。在本文研究的基础上,将研究中采用的自编码器进行分割,取编码器部分连接上单层分类网络,作为分类器开展自监督学习。目前已经在大数据集上取得了不错的效果,但针对多组学这类小样本、高维度数据集,还需要时间进一步调试与研究工作。
总之,基于深度自编码器的多组学整合可以增强人们对于癌症分子亚型的理解,有助于探索癌症亚型分类新方法,并能帮助医疗人员制定个性化的治疗策略。
猜你喜欢
网络安全与数据管理(2022年1期)2022-08-29
四川大学学报(自然科学版)(2021年6期)2021-12-27
皮肤病与性病(2021年3期)2021-07-30
科学技术创新(2021年5期)2021-03-17
计算机应用(2020年12期)2020-12-31
——编码器
演艺科技(2020年7期)2020-08-13
中国动物检疫(2020年1期)2020-01-08
中国兽药杂志(2019年4期)2019-05-15
探测与控制学报(2015年4期)2015-12-15
