
骨质疏松症中骨相关细胞自噬的作用及其靶向治疗
2022-08-30 05:58薛海鹏李志超吴燕徐展望
中国骨质疏松杂志 2022年8期
薛海鹏 李志超 吴燕 徐展望
1. 山东中医药大学,山东 济南 250014
2. 山东中医药大学附属医院,山东 济南 250014
3. 山东第一医科大学第一附属医院,山东 济南 250014
骨质疏松症(osteoporosis,OP)是一种常见的骨骼疾病,给卫生保健和社会带来巨大负担。其特点是由于绝经、衰老和相关药物不良反应等多种因素引起的骨量和微结构丧失,导致骨组织退化和骨脆性增加[1]。一项基于我国居民OP流行病学调查[2]结果显示,50岁以上人群OP患病率为19.2 %,65岁以上人群的患病率则高达32.0 %,已经成为我国的重要健康问题之一。OP的发生发展与骨内稳态异常变化息息相关,骨内稳态主要由骨髓基质细胞(bone marrow mesenchymal stem cells, BMSCs)分化而来的成骨细胞(osteoblasts,OBs)与破骨细胞(osteoclasts, OCs)激活同步的复杂机制维持,从而将骨形成与骨吸收耦合起来[3]。在正常情况下,骨形成与骨吸收的动态平衡使骨代谢保持稳定,当平衡被打破时,就可能造成骨内稳态异常变化,最终导致OP发生。自噬是一种应激反应性的分解代谢过程,在维持细胞和组织稳态中起着关键作用。越来越多的证据[4]表明,自噬与骨稳态之间的重要关联是由BMSCs、OBs、OCs介导的,自噬在OP发病机制中发挥着重要作用。本文拟对OP与自噬之间相关关系的研究展开探讨,旨在更好地理解这两个过程之间的相关性,并总结基于细胞自噬防治OP的不同方式以及面临的挑战。
1 骨相关细胞自噬与骨质疏松症
自噬发生在每个细胞中,是维持细胞活力的基础机制,其过程分为自噬启动、自噬体延长和成熟、自噬体降解和回收几个阶段,这一动态循环过程被称为自噬通量,在维持细胞内稳态中发挥重要作用。骨骼功能作用的发挥需要骨组织的长期稳定,在BMSCs、OBs、OCs协同作用下,骨组织不断被重塑。骨重塑对于微骨折的修复以及骨骼对不同机械刺激的适应和钙稳态是必要的,这一过程中的任何不平衡,使骨再吸收超过形成,都可能导致骨丢失和OP的发生[5]。而自噬与BMSCs、OBs、OCs增殖分化、功能发挥密切相关,自噬水平的变化关系着骨内稳态的变化,被认为是OP发病的关键因素。
1.1 自噬在BMSCs中的作用
BMSCs具有分化为多种骨相关细胞的能力,其异常分化在OP发病机制中起着关键作用[6]。氧化应激是由活性氧(reactive oxygen species, ROS)过度生成引起的,过度氧化应激造成BMSCs衰老和凋亡,抑制成骨分化,导致骨形成缺陷[7]。而提高自噬水平可通过拮抗细胞凋亡来抑制ROS诱导的细胞死亡,延缓BMSCs衰老,维持其干性[8]。同时,BMSCs分化需要不断增加的能量,自噬降解产物可作为细胞代谢的可选营养源循环利用。未分化BMSCs存在高水平自噬活性,在早期成骨分化过程中,BMSCs胞体内也有大量自噬体聚集,表明自噬是BMSCs分化过程中一个有效能量补给途径[9]。而老年BMSCs更容易分化为脂肪细胞而非OBs,其自噬活性降低,使用自噬抑制剂3-MA可减少BMSCs向OBs分化和增殖,而激活剂雷帕霉素则可逆转这一过程[10]。说明通过调整自噬,恢复BMSCs成骨分化能力可以有效减少OP患者骨丢失,这为OP提供了一项潜在治疗策略。
1.2 自噬在OBs中的作用
OBs来源于BMSCs,其数量或活性失调与OP等骨疾病的病理生理机制相关,而自噬对OBs各项功能均具有重要影响。一方面,自噬可作为OBs的细胞保护反应,通过降解受损细胞器保护OBs免受细胞毒性刺激。氧化应激是OBs骨形成受损的重要原因,自噬的激活限制OBs内ROS水平并消除受损线粒体,挽救细胞凋亡[11]。并且,OBs中的自噬也在酸性pH[12]和炎症环境[13]等刺激下触发,以防止细胞凋亡。另一方面,自噬调节OBs参与的骨形成和重塑作用。ATG5、ATG7和Beclin-1是自噬过程的必需基因,在体外,ATG7和Beclin-1缺乏显著降低OBs矿化[14],OBs中ATG5缺失会降低体外矿化能力,并导致体内骨体积减少[14-15]。同样,另一项研究发现[16],沉默ATG5可抑制OBs增殖和分化,并且更容易受到氧化应激损害,而ATG5过表达增强OBs抗氧化能力,减少细胞凋亡。这说明自噬介导BMSCs向OBs分化的过程,并且调控OBs活性、凋亡、增殖、分化以及骨基质矿化,在维持骨微结构和内环境稳定中发挥重要作用。
1.3 自噬在OCs中的作用
OCs来源于单核造血系,主要受巨噬细胞集落刺激因子(monocyte colony-stimulating factor,M-CSF)和核因子κB配体受体激活剂(receptor activator for nuclear factor-κB ligand,RANKL)调控,两者在功能性OCs形成中起着关键作用。在破骨前体细胞成为OCs的过程中,Beclin-1通过自身泛素化促进RANKL刺激OCs进行分化[17]。OCs骨吸收功能是通过含有分泌型溶酶体和肌动蛋白环的褶皱缘来实现的。肌动蛋白环由足小体形成,褶皱缘可以通过分泌组织蛋白酶K(cathepsinK,CTSK)降解骨基质,自噬参与骨吸收过程中肌动蛋白环和褶皱边缘的形成。在表达CTSK的细胞中,Beclin-1敲除会损害OCs功能,导致皮质骨厚度增加[17]。CTSK受ATG5调节,ATG5缺失影响溶酶体关联膜蛋白1进入肌动蛋白环的过程,使得CTSK减少,骨吸收能力受损,而ATG7敲除同样影响OCs骨吸收功能[18]。蛋白激酶3(Kindlin-3,K3)是足体中必需的接头蛋白,其缺失导致OCs足体环功能障碍,K3可以与自噬相关基因LC3Ⅱ相互作用,来调节OCs迁移[19]。以上表明自噬相关基因ATG5、ATG7、Beclin-1、LC3Ⅱ均参与到OCs活动,对OCs分化、迁移、骨吸收功能起到重要调控作用。
2 调控自噬治疗骨质疏松症
调控自噬治疗骨质疏松症的相关策略见图1。
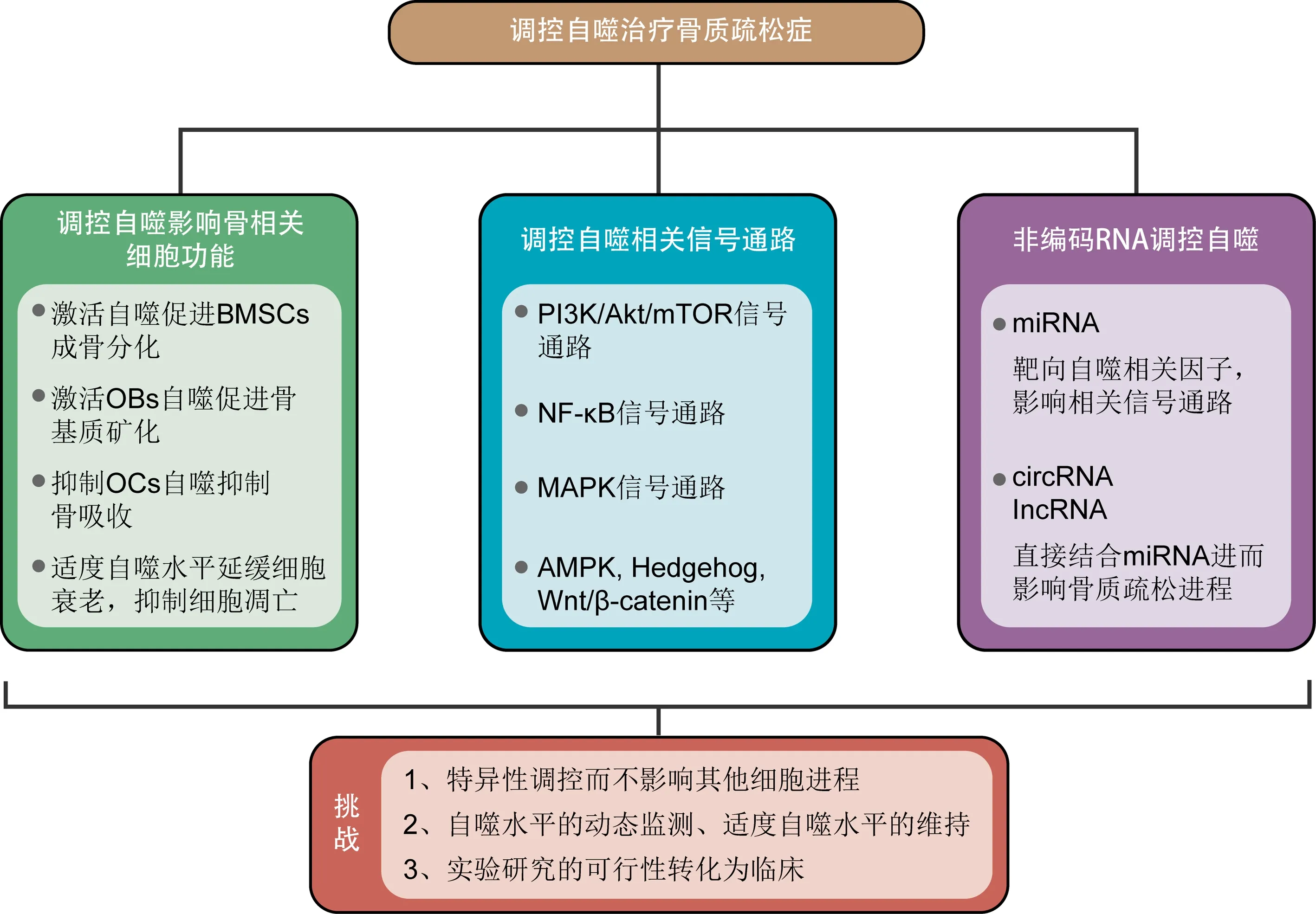
图1 不同方式干预自噬治疗骨质疏松症策略图
2.1 调控自噬影响骨相关细胞功能
2.1.1调节自噬促进BMSCs成骨分化:BMSCs功能在OP中起着关键作用,老年BMSCs分化为OBs的能力下降,这种变化导致骨形成减少进而导致OP,因此BMSCs的精细调控对维持骨内稳态起着重要作用[20]。通过激活自噬促进BMSCs成骨分化有利于骨重塑中骨形成增加,协调成骨成脂分化平衡。最近的研究[21]发现,清道夫受体Scara3通过促进Foxo1表达和自噬来促进OVX小鼠骨形成,减少骨髓脂肪积累,并减轻老年小鼠骨丢失,提示通过靶向Scara3来调节脂肪细胞与OBs分化之间的转换,是骨丢失和骨质疏松的潜在治疗靶点。Vidoni等[22]研究发现白藜芦醇和成骨诱导因子通过AMPK/Beclin-1途径协同激活自噬诱导人牙龈间充质干细胞的成骨分化,表明自噬调节是促进人牙龈间充质干细胞成骨分化的重要手段。
2.1.2调节OBs自噬促进骨基质矿化:OBs参与的矿化过程是骨组织发育和代谢中的重要步骤,骨矿化程度也决定了骨的抗压缩能力的强弱,矿化异常可导致OP等多种疾病。恢复OBs自噬水平可以消除错误折叠的骨基质蛋白,提高钙磷结晶运输效率,促进矿化基质产生。最近,一些药物被证明用于调节OBs的自噬水平来促进骨基质矿化。例如,熊果苷可改善地塞米松(dexamethasone,Dex)诱导的小鼠OP,通过激活自噬促进OBs分化和矿化,抑制骨吸收,促进骨形成[23]。硅酸盐纳米粒子C2S NPs通过激活自噬,进而触发wnt/β-catenin通路来促进OBs分化和矿化能力[24]。
2.1.3调节OCs自噬抑制骨吸收:由于OCs数量和活性增加,OCs骨吸收增强,导致骨结构受损和骨量降低,这是OP等骨疾病的共同特征,因此调整自噬抑制OCs分化和骨吸收可能是防治骨丢失的有效手段。例如,丹参-葛根能够通过调节自噬和氧化应激介导的OCs分化来改善OP[25]。熊果酸能够通过抑制自噬介导的OCs分化,改善骨小梁结构恶化,降低血清OCs特异性细胞因子表达,对卵巢切除术(OVX)大鼠骨质疏松有明显的保护作用,并减轻肾脏损害[26]。
2.1.4延缓细胞衰老、抑制细胞凋亡:衰老是生物体不可避免的,但是通过延缓衰老、预防和治疗某些衰老相关疾病如衰老性OP,是完全可行的。在衰老细胞中观察到自噬水平下降和溶酶体功能障碍,因此,药物干预自噬可能是一种前瞻性抗细胞衰老的方法。例如在衰老骨组织中,雷帕霉素通过激活骨细胞自噬,减少年龄相关的骨细胞凋亡,使骨吸收和骨形成达到平衡[27]。同样,激活自噬以维持骨相关细胞活性和防止凋亡是治疗OP的有效方法。例如芍药苷能够通过调节Akt/mTOR/自噬信号通路,抑制Dex诱导的OBs凋亡,促进细胞活力和骨代谢[28]。
2.2 调控自噬相关信号通路
2.2.1PI3K/Akt/mTOR信号通路:PI3K/Akt/mTOR信号通路与细胞生长、增殖、寿命等直接相关,目前该通路在OP中调控自噬的研究最为深入,但仍存在一些疑惑和矛盾。一方面,部分研究[29]通过抑制PI3K/Akt/mTOR,诱导自噬介导BMSCs向OBs分化,促进OBs增殖和矿化,减轻骨细胞凋亡。例如在阿尔茨海默病和OP相关性研究[30]中发现,阿尔茨海默病典型病理产物淀粉样蛋白-β1-42作为外部刺激直接抑制BMSCs增殖,而抑制Akt/mTOR通路激活自噬可逆转这一效果。另一方面,也有研究[31]发现糖皮质激素通过抑制PI3K/Akt/mTOR通路诱导体内OCs自噬增强从而导致骨丢失,而激活该通路则可逆转OCs自噬。这反映出三个问题:一是自噬在OCs中的作用仍不完全明确;二是应该选择抑制该通路诱导自噬从而促进成骨,还是激活该通路抑制自噬从而减轻骨丢失;三是如何调节特定细胞自噬并将这些可行性转化为临床。
2.2.2NF-κB信号通路:NF-κB蛋白由p65和p50亚单位构成二聚体,并与抑制蛋白IκB结合构成三聚体复合物而处于失活状态,当刺激因素激活NF-κB后,磷酸化IKKβ亚基,IκB被置换出来并受到蛋白酶体降解,从而活化NF-κB并使其易位进入细胞核。被激活的NF-κB信号通路可以促进OCs形成与活化,从而增加骨吸收[32],而NF-κB的失活促进OBs体外分化和体内骨形成[33]。NF-κB亦是典型促炎信号通路,有研究发现[34]ω-3游离脂肪酸通过增强自噬降解炎性复合物从而减轻炎症,也可抑制NF-κB活化来阻止炎症启动,提示NF-κB、自噬与炎症之间存在着潜在的相互联系。
2.2.3MAPK信号通路:MAPK信号通路包含4条分支路线,即ERK、JNK、p38和ERK5。MAPK/ERK通路正调节自噬,通过提高自噬保护氧化应激下的骨细胞[35],并参与破骨前体细胞增殖和OCs凋亡[36]。MAPK/JNK通路常与RANKL信号通路交联,参与OCs形成、分化、凋亡过程[37]。MAPK/p38通路则对OCs分化和骨吸收起调控作用[38]。目前对于MAPK/ERK5通路研究相对匮乏,但明确的是M-CSF可通过诱导ERK5磷酸化来参与OCs分化过程[39]。MAPK通路参与OP中自噬的作用机制复杂,在许多研究中仍存在争议,如ERK信号通路对BMSCs成骨分化起正向[40]还是负向[41]作用仍然存疑,而且MAPK通路分支功能广泛,多条通路之间交联机制仍未清晰,因此要思考如何在多条通路中起到特异性调控作用。
2.2.4其他信号通路:除上述信号通路外,AMPK信号通路[42]、Hedgehog信号通路[43]、Wnt/β-catenin信号通路[44]等均介导自噬参与骨内稳态调节,各信号通路间交联复杂,其潜在作用机制仍需不断研究。
2.3 非编码RNA调控自噬
非编码RNA(ncRNA)包括miRNA、lncRNA和circRNA等,在细胞增殖、分化、凋亡和自噬等过程中起着重要调节作用。Jin等[45]基于Ilumina深度测序技术鉴定了260个circRNA、70个lncRNA和13个miRNA在绝经后OP患者和健康对照组中差异表达。自噬和miRNA、lncRNA、circRNA都参与骨代谢,而为了挖掘ncRNA与自噬相互关系在OP中的治疗作用,需要分析两者之间的交互作用对骨稳态的影响。有关miRNA与自噬在OP中的关系多体现在miRNA对自噬相关因子或信号通路的调控作用。例如,miRNA-20a可通过靶向RAW264.7细胞中HIF-1α显著下调自噬标志物(LC3、p62、Atg5、Atg12、Atg7、Beclin-1等),这是缺氧诱导的OCs分化的关键机制[46]。miRNA-378通过影响PI3K/Akt通路降低OBs活力和矿化能力[47]。lncRNA和circRNA都可以通过直接与miRNA结合而影响OP进程,并与自噬密切相关,但目前有关circRNA和lncRNA的研究还比较表浅。Ji等[48]研究发现,circRNA 002682通过miRNA-188-3p靶向上调Beclin-1介导的自噬,从而促进OBs分化。Wu等[49]研究发现,lncRNA ZFAS1通过上调miRNA-499上调EPHA5表达从而抑制BMSCs成骨分化,且ZFAS1敲除会促进成骨分化,抑制脂肪分化,促进细胞自噬,抑制细胞衰老,表明ZFAS1/miR-499/EPHA5途径可能在BMSCs成骨和成脂之间的相关转换中起重要作用。ncRNA在骨组织中的生物学作用至关重要,针对其与自噬的相互作用来治疗OP在临床中的应用潜力无疑是巨大的,ncRNA/自噬模拟物或ncRNA/自噬抑制剂的研发可能是有效预防骨丢失的治疗方法。
3 讨论与展望
自噬在OP中扮演着复杂的角色,在不同类型、程度的OP中,自噬的抑制或激活程度、功能均不同。调节自噬的方式也是复杂多样的,从自噬影响不同骨相关细胞的功能、自噬相关信号通路的信息传递、非编码RNA与自噬的相互作用等多种方式,均能使合适的自噬水平参与骨内稳态调节,进一步影响OP发生发展。但显然多种方式又是互相交联的,特异性靶向调节而不影响其他细胞过程仍然存在挑战。骨重塑过程是动态的,自噬也是动态的,提示需要更好的监测自噬与OP的动态关系。这无疑是困难的,但对于加快推进分子机制的研究和药物的研发具有极大的指导意义。
猜你喜欢
水土保持学报(2022年5期)2022-10-10
科学技术与工程(2022年22期)2022-09-29
大电机技术(2022年4期)2022-08-30
农业工程学报(2022年10期)2022-08-22
农业机械学报(2022年7期)2022-08-08
大电机技术(2022年3期)2022-08-06
上海师范大学学报·自然科学版(2022年3期)2022-07-11
耕作与栽培(2022年2期)2022-07-01
中国美容医学(2020年6期)2020-06-29
医学信息(2020年4期)2020-04-09
