
聚乙二醇对聚乳酸/淀粉纳米晶复合材料性能的影响
2022-08-25 08:04曲玉婷王立梅
中国塑料 2022年8期
曲玉婷,王立梅 ,齐 斌
(1.苏州大学药学院,江苏 苏州 215000;2.苏州市食品生物技术重点实验室,江苏 常熟 215500;3.常熟理工学院生物与食品工程学院,江苏 常熟 215500)
0 前言
PLA又称聚羟基丙酸或聚交酯,是由乳酸(LA)单体缩聚而成的可生物降解高分子材料[1]。因具有良好的力学、光学和物理性能,使其在全球生物基塑料的生产中占比最大[2‐3]。但是,PLA具有脆性大、结晶速率慢、生产成本高等缺点,限制了其应用范围[4]。为克服以上缺点,通常需要对PLA进行改性,其中,共混改性是最简单有效的方法之一。SNC是1种淀粉经水解后产生的结构均匀的刚性纳米粒子,由于具有晶体结构致密、比表面积和表面能大等特点,将其均匀分散在聚合物中可以改善复合材料性能[5‐6]。然而,SNC上存在的大量羟基限制其在PLA等疏水聚合物中的均匀分散,从而导致SNC与PLA之间的界面结合能力不佳[7]。PEG是1种安全无毒、可生物降解的高效增塑剂,与PLA共混时表现出良好的相容性和塑化能力[8‐9]。除此以外,PEG还被发现可以降低PLA基体与填料之间的界面能,增加基体与填料之间的界面结合力,从而提高复合材料性能[10‐12]。
目前,增加PLA和SNC界面相容性的方法主要是对SNC进行表面修饰,用疏水官能团取代羟基,增加SNC与PLA的相容性。Zamir等[13]利用酯化反应将在SNC上接枝LA,接枝后SNC与PLA基体的界面黏附性和相容性得到明显改善。Takkalkar等[14]制备了SNC和乙酰化SNC用于增强PLA,乙酰化SNC在PLA中具有更好的分散性,与聚合物基体之间的相互作用得到增强。本文以PLA为基体,SNC为填料,采用共混法制备PLA/SNC复合材料,探究PEG的添加对PLA/SNC复合材料结晶行为、力学性能以及界面相容性的影响。
1 实验部分
1.1 主要原料
PLA,4032D,美国Natureworks公司;
SNC,100 nm,自制;
PEG,分子量2 000,国药集团化学试剂有限公司;
氯仿,分析纯,江苏强盛功能化学股份有限公司。
1.2 主要仪器和设备
磁力加热搅拌器,RCT basic,德国IKA公司;
DSC,Pyris 1 DSC,美国PerkinElmer公司;
徕卡超薄切片机,EM UC7,德国徕卡公司;
PLM,ECLIPSE 50i POL,日本Nikon公司;
XRD,D/max 2500 PC,日本理学株式会社;
拉伸试验机,WDT‐5,深圳市凯强利实验仪器有限公司;
SEM,JSM‐6360LA,日本电子株式会社。
1.3 样品制备
将SNC和PEG加入到适量的氯仿中,超声分散;称取一定量的PLA加入分散有SNC和PEG的氯仿中,室温下搅拌3 h,静置一段时间以消除溶液中的气泡;将混合溶液倒入聚四氟乙烯模具中,放入通风橱中室温烘干过夜,将薄膜剥离后放入60℃烘箱中烘干,得到厚度为(90±5)μm的PLA及其复合材料;复合材料配比如表1所示。
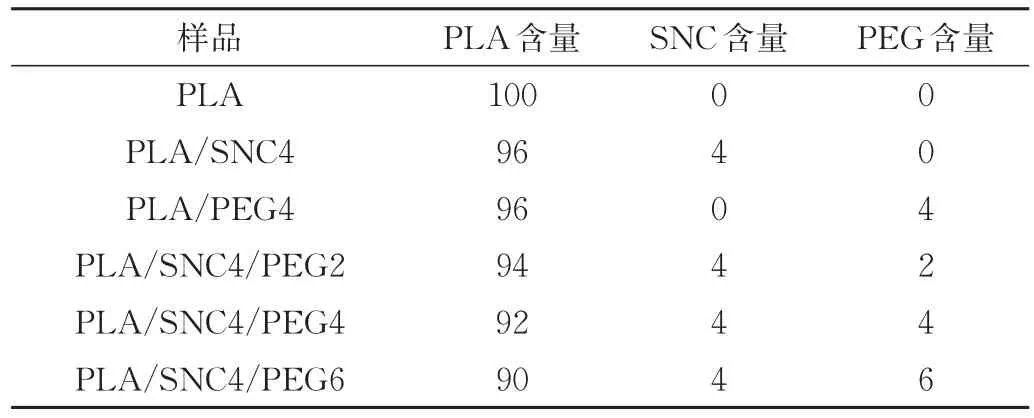
表1 PLA复合材料的配方 份Tab.1 Formula of PLA composite materials phr
1.4 性能测试与结构表征
热性能分析:采用DSC对PLA及其复合材料的结晶行为进行测试;取试样8~12 mg,氮气保护环境下进行以下热循环;首先从室温快速升温至200℃,并恒温5 min以消除热历史,然后以不同的降温速率(1、2.5、5、10℃/min)冷却至30℃并记录结晶放热曲线,得到PLA复合材料的结晶度(Xc,%)和结晶峰温度(TP),其中Xc由式(1)计算:

式中 ΔHC——结晶焓,J/g
ΔH——完全结晶放热焓,为93.3 J/g
ω——复合材料中PLA的质量分数,%
球晶形态分析:采用徕卡超薄切片机将材料切成厚度10 μm左右的薄片,将其置于2片圆形盖玻片中央,在PLM热台上快速升温至200℃至充分熔融,并恒温1 h以消除热历史,然后在氮气环境下以0.5℃/min冷却至110℃观察球晶形态并拍照;
球晶结构分析:采用XRD进行表征,测试条件为Cu‐Kα射线,Ni片滤波,波长0.15 mm,辐射管电压40 kV,管电流100 mA,扫描速率0.02°/0.2 s,衍射角度2θ为10°~40°;
力学性能分析:利用拉伸试验机在室温条件下参照GB/T 1040.1—2018进行测试,试样标距25 mm,拉伸速率为5 mm/min,每组试样测试4次取平均值;
形貌分析:将制备的复合材料试样使用液氮脆断,其断面经喷金处理后采用SEM观察断面形貌,放大倍率为104倍,加速电压15 kV。
2 结果与讨论
2.1 非等温结晶行为分析
图1为PLA/SNC4/PEG4复合材料在不同降温速率下(1、2.5、5、10℃/min)的非等温结晶曲线,从图中可以看出,随着降温速率的增大,复合材料的结晶峰逐渐变宽,TP逐渐向低温移动。当降温速率为10℃/min时,几乎看不到结晶峰。这是因为结晶过程具有热滞后性,PLA分子链段运动进入晶格是1个松弛的过程,需要一定的时间来完成,使得在降温过程中结晶会发生滞后,并且随着降温速率的增加滞后也会相对延长。当降温速率较慢时,分子链有足够的时间排列活动,晶核能够在较高温度下形成,TP更高,因此形成的结晶较为完善。而当降温速率越快时,更多的分子链要在低温下进入晶格形成晶体,由于在低温条件下分子链的运动速度较慢,使得滞后越多,从而导致结晶温度范围越宽,结晶的完善程度越低[15]。
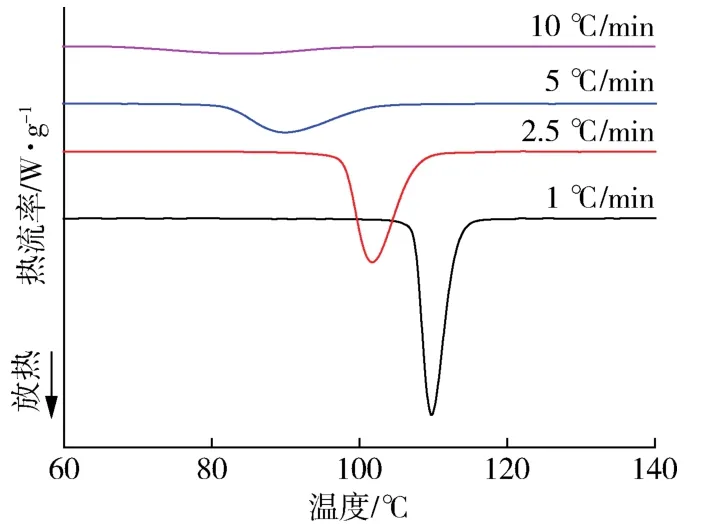
图1 PLA/SNC4/PEG4复合材料在不同降温速率下的结晶放热曲线Fig.1 DSC cooling exotherms of PLA/SNC4/PEG4 composite materials at different cooling rates
图2为降温速率为1℃/min时PLA及复合材料的结晶放热曲线。从图中可以看到,在该测量条件下,PLA没有明显的结晶峰,说明PLA为无定形状态,在此降温条件下几乎不结晶,这与Courgneau的结果一致[16],而PLA/PEG4与PLA/SNC4复合材料的结晶峰较为明显。同时,与PLA/PEG4以及PLA/SNC4复合材料相比,PLA/SNC4/PEG4复合材料的结晶峰更窄,TP更高,结晶速率也更快。表2为PLA及复合材料在不同降温速率下的TP和Xc。从表中可以看出,在降温速率为1℃/min时,PLA/PEG4复合材料的TP为89.7 ℃,Xc为20.50%,由此可见,PEG作为增塑剂,在一定程度上可以增加PLA分子链的运动能力,从而提高其TP和结晶速率[17]。PLA/SNC4复合材料的TP为105.5℃,Xc为27.98%,说明在PLA/SNC复合体系中,SNC通过异相成核的作用使得PLA的结晶速率和TP有所提高。当二者同时添加时,PLA/SNC4/PEG4复合材料的TP约为109.6 ℃,Xc为30.18%。并且在不同的降温速率下,PLA/SNC4/PEG4复合材料的Xc和TP均比PLA、PLA/SNC以及PLA/PEG复合材料要高。这是因为PLA/SNC4/PEG4复合材料一方面可以通过SNC的异相成核作用增加晶核数量,提高成核速率;另一方面可以通过PEG提高PLA的链迁移率,从而提高晶体的生长速率,在二者的协同作用下,使得复合材料的结晶速率大大提高。

图2 样品在降温速率为1℃/min下的结晶放热曲线Fig.2 DSC cooling exotherms of the samples at a cooling rate of 1℃/min
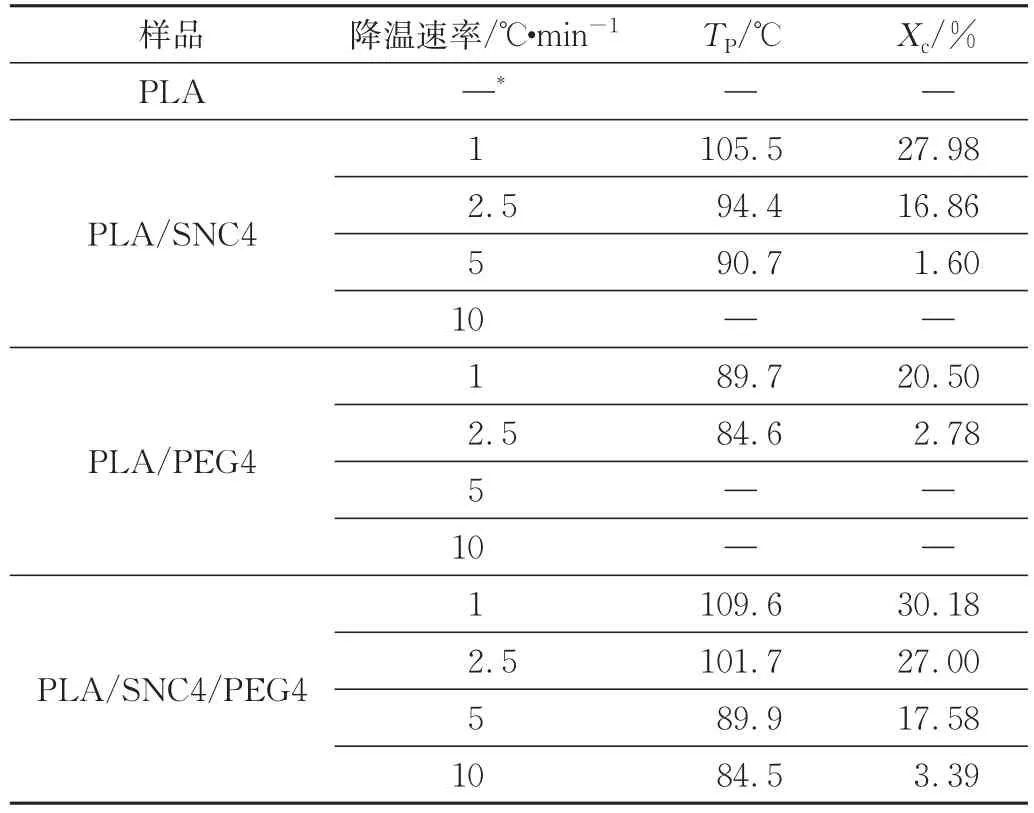
表2 样品在不同降温速率下的TP和XcTab.2 TPand Xcof the samples at different cooling rates
2.2 球晶形态分析
图3为PLA及其复合材料的球晶形貌。图3(a)为PLA的PLM照片,可以看到明显的黑十字消光现象,且晶粒尺寸较大,晶粒密度较低,表现为球晶的特征。图3(b)是PLA/PEG4复合材料的PLM照片,在加入PEG后,可以看到球晶尺寸变小,晶粒密度有所增加。这是因为PLA的分子间作用力强,链段运动能力弱,而PEG的加入削弱了PLA分子链间的氢键作用,增加了PLA分子的链段运动能力,从而改善其结晶能力。图3(c)为PLA/SNC4的PLM照片,加入SNC后晶粒尺寸明显变小,晶粒密度变得很大。这是因为SNC发挥了异相成核作用,在PLA结晶过程中提供了更多的晶核,PLA分子链可以附着在SNC上结晶生长。同时由于晶核较多导致球晶之间相互碰撞抑制了进一步的生长,使得晶粒的尺寸较小。此外,由于PLA/SNC4形成的球晶尺寸小且密度大导致其重叠生长,球晶边缘模糊。图3(d)为PLA/SNC4/PEG4复合材料的PLM照片,可以看到与PLA/SNC4和PLA/PEG4复合材料相比,其球晶的尺寸更小,密度更大。这可以解释为,一方面SNC的存在提供更多的晶核,PEG的存在增加了PLA分子的链段运动能力,二者协同大大提高了PLA/SNC4/PEG4复合材料的结晶能力;另一方面PEG的存在提高了SNC在PLA中的分散程度,使得SNC更好地发挥异相成核的作用。此外,在照片中还呈现许多气泡,这可能是由于结晶速度过快导致体积迅速收缩,从而形成气泡。
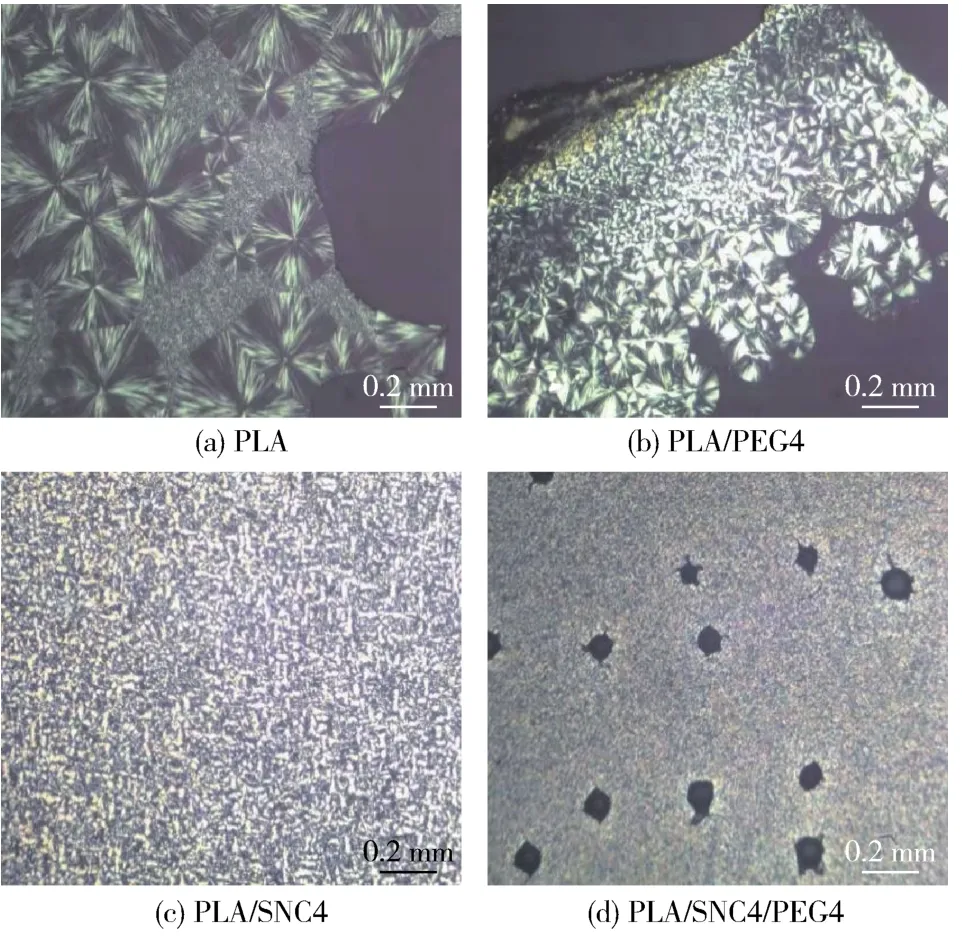
图3 样品在110℃下的PLM照片Fig.3 PLM images of the samples at 110℃
2.3 结晶结构分析
图4为PLA及其复合材料的XRD谱图。从图中可以看到,纯PLA呈现非常宽的弥散峰,说明PLA在室温下基本为无定形状态。当加入SNC和PEG后,PLA/PEG、PLA/SNC以及PLA/SNC/PEG复合材料均在2θ=14.5 °、16.4 °、18.7 °、22.0 °处有明显的衍射峰。其中,在2θ=14.5 °处的衍射峰对应(010)晶面的α‐型晶体,在2θ=16.4°处的衍射峰对应(110)或(200)晶面的α‐型晶体,在2θ=18.7 °的衍射峰对应(203)晶面的α‐型晶体,在2θ=22.0 °附近的衍射峰对应(015)晶面的α‐型晶体[18]。由此可见,该复合材料的结晶相为典型的α晶型。当加入PEG后,复合材料衍射峰的位置和数量没有发生变化,仅表现为衍射峰强度的增加,说明PEG的加入不会导致PLA/SNC复合材料晶型的改变。而PLA/SNC4/PEG4复合材料的衍射峰最强,其原因一方面是SNC和PEG对复合材料的结晶速率的促进作用,另一方面可能由于PEG增强了PLA/SNC4复合材料中的氢键,促进了结晶分子的重排[19]。
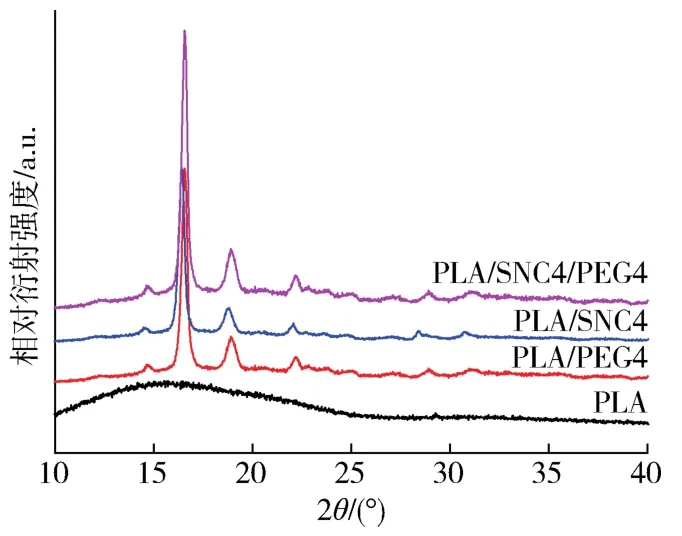
图4 样品的XRD谱图Fig.4 XRD patterns of the samples
2.4 力学性能分析
PLA及PLA复合材料的拉伸强度和断裂伸长率如表3所示。PLA的拉伸强度为45.39 MPa,PLA/SNC4复合材料的拉伸强度为47.22 MPa。复合材料拉伸强度的提高可能是因为SNC为刚性材料,具有较高强度和弹性模量,同时,SNC能够发挥异相成核的作用,有效提高PLA的结晶速率和Xc,增强PLA的分子间作用力,从而提高其拉伸强度。PLA的断裂伸长率为8.70%,添加SNC后,PLA/SNC4复合材料的断裂伸长率有所下降。这是因为PLA本身为脆性材料,而SNC的高模量和高强度使得PLA的Xc有所增加,导致复合材料的脆性增加。同时,SNC和PLA之间的界面相容性差,使得复合材料的应变能力减弱,断裂伸长率下降。加入PEG后,PLA/PEG4复合材料的拉伸强度下降到39.32 MPa,断裂伸长率提高至15.93%,这是因为PEG在进入PLA大分子链后起到了稀释作用,降低了PLA大分子间的作用力,使其松弛过程延长,PLA的柔韧性得到了提高[20]。当在PLA/SNC4复合材料中添加不同含量的PEG时,可以看到PLA/SNC4/PEG2复合材料的力学性能最佳,拉伸强度由47.22 MPa提高至47.86 MPa,断裂伸长率提高了约30%。随着PEG含量的增加,PLA/SNC4/PEG复合材料的拉伸强度不断下降。拉伸强度的提高可以解释为,共混物力学性能主要是由共混物的相尺寸和界面强度决定的,SNC在PEG的包裹下提高了在PLA中的分散性,PEG分子链在PLA和SNC中形成缠结,促进了二者之间的相互作用,从而提高共混界面的强度。除此以外,PEG的添加导致SNC/PLA的质量分数增加,使得SNC更好地发挥增强作用。同时,PEG对PLA还有一定的增塑作用,使得PLA/SNC4/PEG2复合材料的拉伸强度和断裂伸长率均得到了提高。而当PEG含量增加时,SNC/PLA的表3质量分数进一步增大。因此,其拉伸强度的降低一方面可能是SNC的相对增加以及PLA的减少使得部分SNC聚集,导致分子间作用力减弱;另一方面是过量的PEG对复合材料的增塑作用大于其微弱的增容作用,最终使得复合材料的拉伸强度下降。

表3 样品的力学性能Tab.3 Mechanical properties of the samples
2.5 断面形貌分析
图5为PLA、PLA/SNC4以及PLA/SNC4/PEG2复合材料断面的SEM照片。图5(a)为PLA的断面,可以看出纯PLA具有相当光滑、平整、连续的断裂表面。图5(b)为PLA/SNC4复合材料的断面形貌,当SNC含量为4%时,由于负载量较低,在PLA中基本可以均匀分散,复合材料的断面略微粗糙。图5(c)为PLA/SNC4/PEG2复合材料的断面形貌,可以看到加入PEG后复合材料的断面相对平整,SNC与PEG的相分界处模糊,这是因为PEG可以与SNC之间形成氢键从而减少SNC之间的团聚,提高了SNC在PLA基体中的分散性,同时PEG分子链在PLA和SNC中形成缠结,增加了二者的界面相容性,这一结论与力学测试结果相符合。

图5 样品断面的SEM照片Fig.5 SEM images of fracture surfaces of the samples
3 结论
(1)SNC和PEG均可以改善PLA的结晶速率;当降温速率为1℃/min时,PLA/SNC4复合材料的TP为105.5 ℃、Xc为27.98%;PLA/PEG4复合材料的TP为89.7℃、Xc为20.50%;PLA/SNC4/PEG4复合材料的TP为109.6℃、Xc为30.18%;SNC与PEG的协同作用使得复合材料的结晶速率和Xc达到最大;SNC和PEG均可以降低PLA的球晶尺寸,增加晶粒密度,当二者同时添加时,PLA/SNC/PEG复合材料的球晶尺寸更小,密度更大;PEG和SNC的添加均没有改变PLA/SNC复合材料的晶型;
(2)随着PEG含量的增加,PLA/SNC4/PEG复合材料的拉伸强度先升高后降低,断裂伸长率不断提高;当PEG含量为2%时,PLA/SNC4/PEG2复合材料的力学性能最佳,其拉伸强度由47.22 MPa提高至47.86 MPa,断裂伸长率由7.83%提高至10.2%;少量PEG的加入在一定程度上改善了PLA与SNC之间的界面相容性。
猜你喜欢
化学工业与工程(2022年1期)2022-03-29
疯狂英语·新读写(2021年5期)2021-11-23
小哥白尼(神奇星球)(2020年7期)2021-01-18
金桥(2020年10期)2020-11-26
陶瓷学报(2020年2期)2020-10-27
Coco薇(2017年7期)2017-07-21
大陆桥视野·下(2016年9期)2017-05-08
科技创新与应用(2017年7期)2017-03-27
红领巾·萌芽(2017年1期)2017-02-06
纺织导报(2014年7期)2014-10-30
