基于3个自噬相关微小核糖核酸表达信息学分析构建肺腺癌预后风险评分模型*
2022-08-19 03:23李小燕
成都医学院学报 2022年4期
秦 花,李小燕,何 杰,李 婷
成都医学院第一附属医院(成都 610500)
随着环境污染加剧、职业暴露和吸烟人群增多、人口老龄化及肺癌影像学筛查普及,肺癌患者数量呈逐年上升趋势[1-2]。据统计,2018年世界人口中肺癌新增病例约200万人,直接或间接因肺癌死亡病例150万人,其中70%以上的肺癌都属于非小细胞肺癌[3]。而近年来,非小细胞肺癌中肺腺癌所占比例高于肺鳞癌,居第一位[3-4]。因肺腺癌起病相对较隐匿,早期并无特殊症状,以致部分患者确诊时已错失手术良机,总体的5年生存率低于20%[5]。尽管目前研究[6]已经发现多种分子生物标志可以辅助临床用于肺腺癌的预后判断,但均有一定的局限性和不足。因此,构建一种新的、有效的预测模型有助于肺腺癌患者的预后判断和个体化治疗。
自噬是一种正常的生物过程,它将细胞质中的物质组成转移到溶酶体中,再将其分解为初级成分[7]。自噬的平衡出现异常将导致肿瘤的发生和发展[8]。微小核糖核酸(microRNA,miRNA)也可以通过调节自噬相关的信号通路,干扰肿瘤细胞的增殖周期和凋亡。如在黑色素细胞瘤中,miR-290-295簇能靶向结合ulk1、atg7等多个自噬相关基因,抑制黑色素瘤细胞自噬性死亡,以提高肿瘤细胞的生存力[9]。基因mtor参与调节细胞自噬的过程,也是miR-1271的靶基因。在非小细胞肺癌中,miR-1271与mtor呈负相关,miR-1271通过吸附mtor基因抑制其表达,从而抑制非小细胞肺癌的增殖[10]。
鉴于miRNA在肺癌自噬中的重要作用,本研究通过比较癌症基因组图谱(the cancer genome atlas,TCGA)数据库中肺腺癌与正常肺组织中不同的自噬基因,寻找与自噬基因相关的miRNA,并构建预后风险评分(risk score,RS)模型,探讨运用自噬相关miRNA预后RS模型预测肺腺癌患者预后的可行性,为肺腺癌的精准医疗提供参考。
1 资料与方法
1.1 临床资料
2021年8月从TCGA官方网站(https://gdc-portal.nci.nih.gov/)下载肺腺癌患者的信使核糖核酸(messenger RNA,mRNA)和miRNA测序数据及临床信息,测序数据由Illumina高通量测序平台获得。数据内容涉及肺腺癌患者535例肿瘤组织和59例癌旁组织及相关临床信息。由于生存时间少于30 d的患者可能导致分析结果出现偏差,因此将其删除,最终纳入肺腺癌患者的例数为490例。
1.2 研究方法
1.2.1 自噬相关miRNA的提取 从人类自噬数据库(human autophagy database,HADb)(http://autophagy.lu/)中获得自噬基因232个,通过ActivePerl软件(版本号5.26)提取自噬基因在TCGA的表达谱以及miRNA的表达谱。采用R软件中的“edgeR”包(https://www.R-project.org)对mRNA和miRNA测序表达值进行log2转换,以log2(fold change)的绝对值>2和伪发现率(false discovery rate,FDR)<0.05设定为阈值,差异基因表达分析采用“edgeR”包。绘制热图展现差异表达的自噬基因和miRNA。使用R软件中的“corrplot”包筛选出与自噬基因相关的miRNA,阈值设定为相关系数|r|>0.4,P<0.05,并绘制网络图。
1.2.2 随机分组产生训练集和测试集 使用Excel软件产生随机数,将490例肺腺癌患者均分为训练集和测试集。训练集用来完成学习标本特征分析和模型构建,测试集用来完成内部验证。
1.2.3 单因素Cox回归分析联合LASSO回归分析筛选关键miRNA 为初步筛选出和生存预后相关的miRNA,使用R包“survival”对自噬相关miRNA进行单因素Cox回归分析,筛选出与预后相关的miRNA,并计算风险比。为避免单因素Cox回归分析结果可能有过度拟合的问题,通过R包“glmet”对单因素Cox回归筛选出的结果再次进行LASSO回归分析。本研究使用10折交叉验证法来确定最小λ值,当λ最小时模型最优化。一旦确定了miRNA,应用它们构建基于如下表达式的预后RS模型,表达式如下所示:
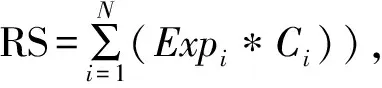
1.2.4 预后模型评价和卡普兰-梅尔估计量法描述生存分析 为验证肺腺癌预后RS模型是否可以用于肺腺癌患者的预后评估,本研究对高风险人群和低风险人群采用卡普兰-梅尔估计量法(Kaplan-Meier,K-M)进行生存分析;其次,再次利用R软件中的“survival”和“time ROC”包绘制3年及5年总生存率的受试者工作特征曲线(receiver operating characteristic curve,ROC)的曲线下面积(area under curve,AUC),评估其预测3、5年生存率的能力。此外,本研究结合TCGA下载的肺腺癌临床相关信息,如年龄、性别、病理分期等,对预后模型进行多因素Cox回归分析,以验证该模型所计算出的危险评估是否可成为独立的预测因子,以诺莫列线图的形式加以展示。
1.2.5 试验验证 1)标本来源:收集成都医学院第一附属医院2016年3月至2019年7月收治的肺腺癌患者50例,相应的50例肺腺癌样本及癌旁正常组织样本通过外科手术或呼吸与危重症医学科纤支镜活检手术获得,病理结果均确诊为肺腺癌。患者及家属签署知情同意书,配合参与本研究试验,伦理申请通过成都医学院第一附属医院伦理委员会批准。2)制剂:Trizol、实时定量PCR所需试剂、RIPA裂解缓冲液(美国,赛默飞世尔科技公司)。3)引物的设计和合成:按照GenBank中提供的人hsa-mir-31,hsa-mir-1293,hsa-mir-548f-1全长基因序列,由杭州艾比肯生物工程(浙江)股份有限公司设计合成,以U6为内参基因,制定的引物序列(表1)。4)PCR实验方法:实时定量PCR严格按照商品的使用说明书进行操作,所获标本进行研磨后,用Trizol试剂从标本中提取总RNA,用ND-1200核酸定量检测仪(美国,赛默飞世尔科技公司)测定提取总RNA的浓度和吸光度值,以吸光度值1.8~2.0为合格。总RNA经过1%琼脂糖凝胶电泳鉴定,所有样本总RNA的吸光度值均为1.8~2.0,提示提取的总RNA质量合格。按照PrimeScriptTMRT Master Mix试剂盒(日本,Takara公司)合成cDNA,以u6为内参;检测在ABI 8000实时定量PCR仪进行,反应条件按照如下标准进行设定:95 ℃预变性30 s;95 ℃、5 s;60 ℃、34 s;总共40个循环;60 ℃退火30 s。计算2-ΔΔct作为相对表达量。

表1 目的miRNA和参照基因的引物序列
1.3 统计学方法

2 结果
2.1 纳入患者的基本情况
符合纳入标准的肺腺癌患者490例,随机分为训练集和测试集,两组患者的临床特征,包括年龄、性别、肿瘤分期、生存状态和生存时间等,差异无统计学意义(P>0.05)(表2),说明训练集和测试集的样本来源于同一个总体,随机分组合理。
表2 纳入患者的基本资料和特征
2.2 自噬基因在肺腺癌组织和癌旁组织的差异表达
总体样本中,经R软件分析,满足条件log2FC的绝对值>2和FDR<0.05的自噬基因共有30个。基因表达谱聚类热图展示了其在样本中的表达(图1)。
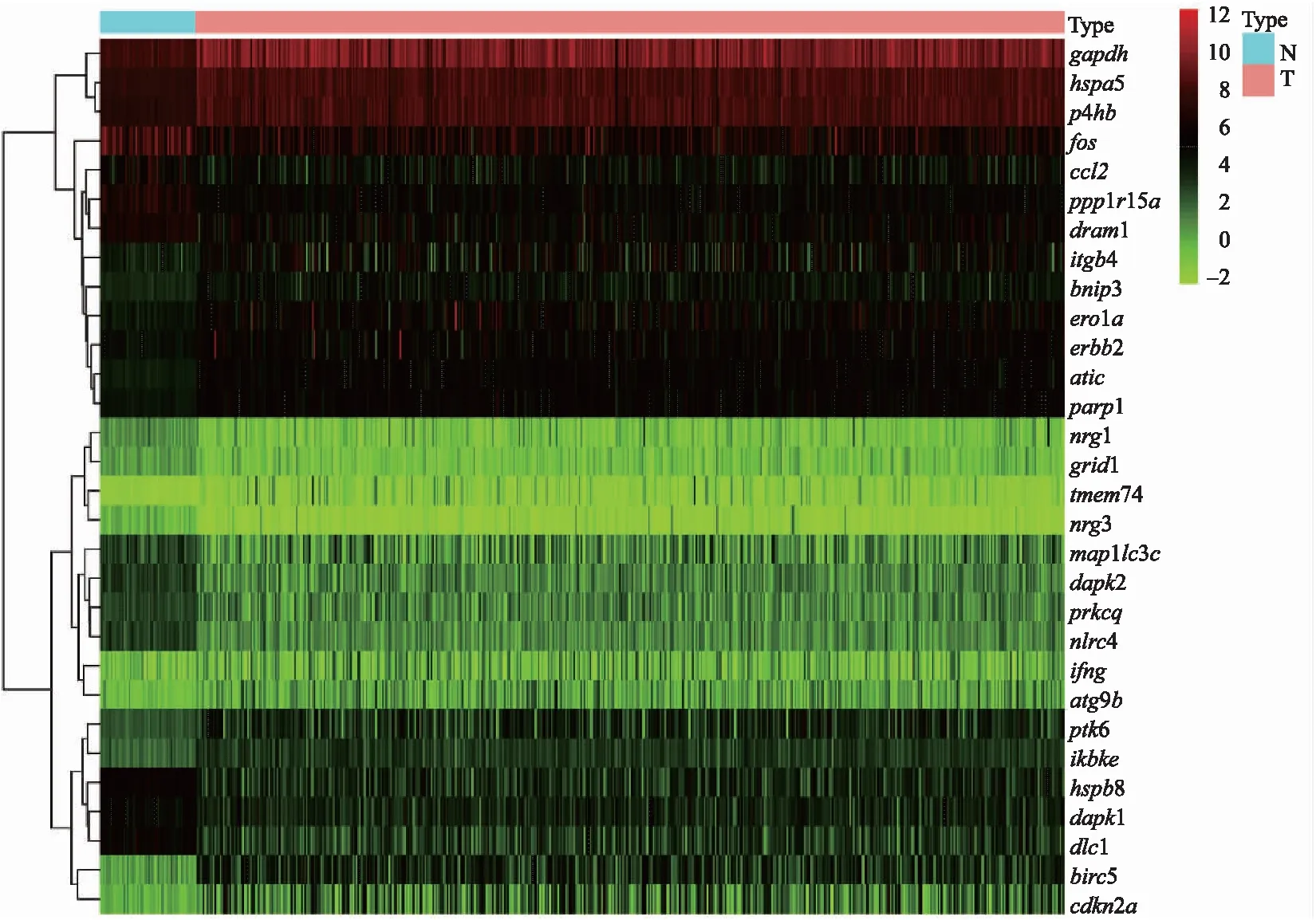
图1 59例正常肺组织和490例肺腺癌组织中30个自噬基因的差异表达
2.3 自噬相关miRNA的确定
通过Perl软件共提取出1 881个miRNA,筛选出70个差异表达的miRNA(图2),依据相关系数(|r|>0.4,P<0.05)获得12个自噬相关miRNA,这12个自噬相关miRNA和5个自噬基因相关(图3)。
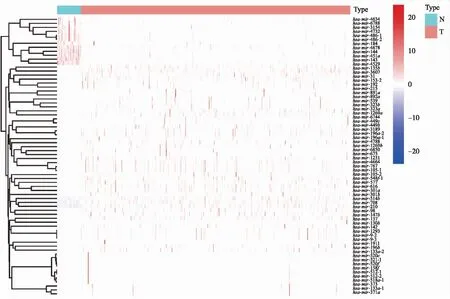
图2 70个差异miRNA的热图
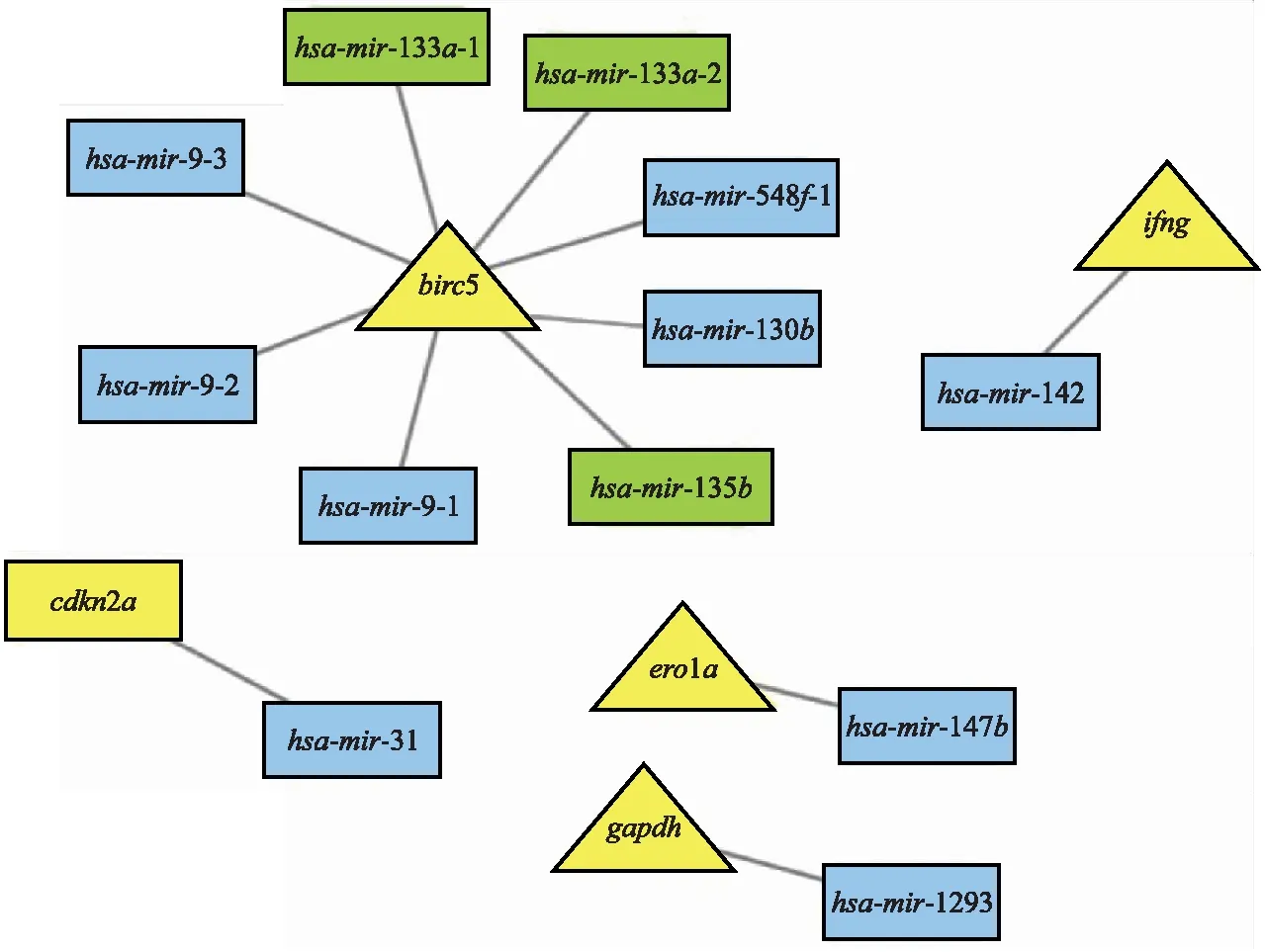
图3 自噬相关的12个miRNA网络互作图
2.4 构建模型
训练集中,采用Perl软件将每个样本的临床信息与miRNA的表达量进行合并,单因素Cox回归分析初步筛选出和肺腺癌预后相关的3个关键miRNA;通过LASSO回归分析进一步确定了3个关键的自噬相关miRNA,分别是hsa-mir-31、hsa-mir-1293和hsa-mir-548f-1(图4)。3个自噬相关miRNA的生物学信息(表3)。
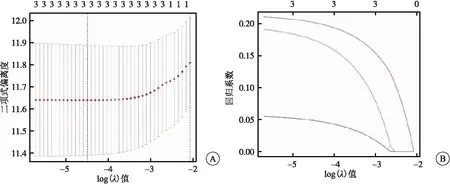
图4 LASSO回归模型筛选变量

表3 3个自噬相关miRNA的详细信息
2.5 肺腺癌预后RS模型构建和评价
训练集中,上述研究中所得的3个自噬相关miRNA,通过RS表达式再次计算出训练集中每个肺腺癌患者的预后RS,RS=0.048×hsa-mir-31+0.201×hsa-mir-1293+0.174×hsa-mir-548f-1,以中位值为界线将患者分成高、低风险组,并构建相应的预后模型。K-M生存分析显示,高风险组患者的中位生存期是1.63年,预测3、5年患者的生存率分别为49%、25%,低风险组中位生存期是2.08年,3、5年生存率分别为74%、52%,低风险组总体生存时间(overall survival,OS)较高风险组长(P<0.05)(图5A);3年总生存率的AUC=0.796,5年总生存率的AUC=0.837(图5B),该预后模型的C指数为0.811。与训练集一样,测试集中高风险分数越高的患者预后越差(P<0.05)(图6A),3年总生存率的AUC=0.684,5年总生存率的AUC=0.646(图6B),并且R软件计算出该预后模型的C指数为0.761。
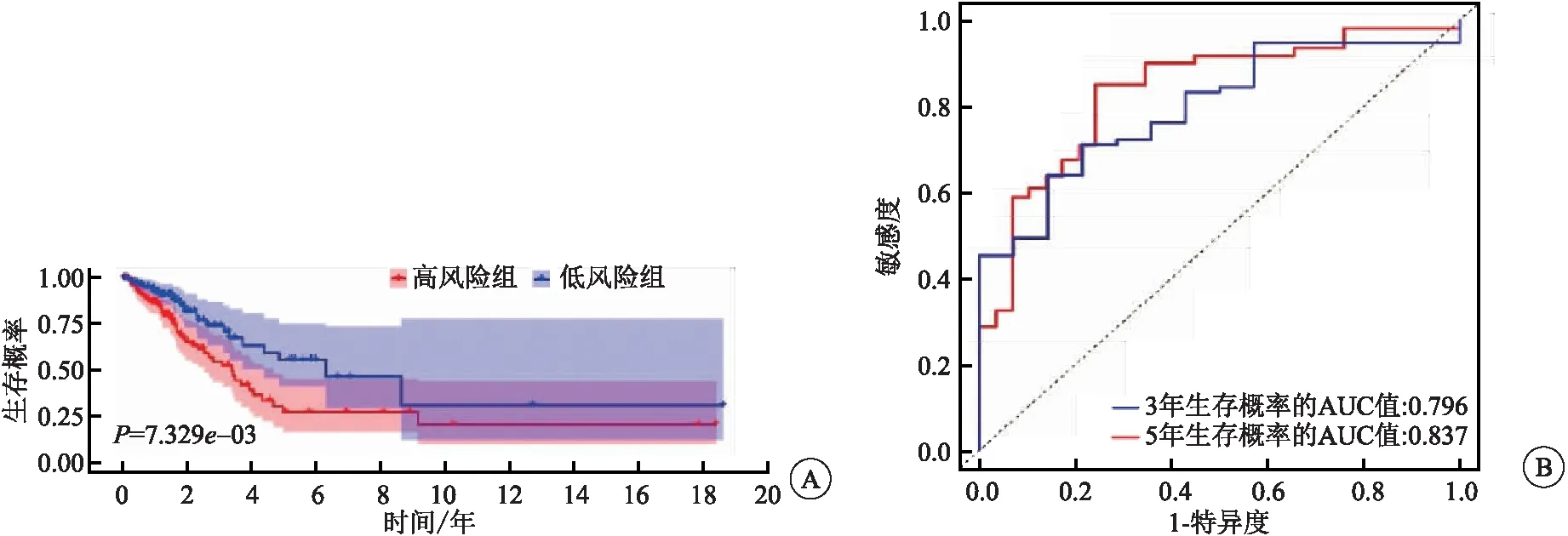
图5 训练集中模型的预测能力评估
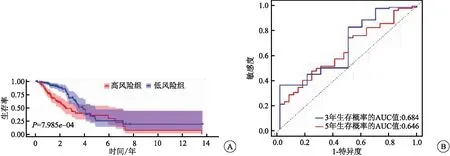
图6 测试集中模型的预测能力评估
2.6 肺腺癌预后RS模型在测试集中的验证
训练集中,以OS为因变量,以RS、年龄、性别、病理分期等多个因素作为协变量进行单因素和多因素Cox回归分析,结果显示RS与患者OS呈负相关,且可作为1个独立的预测预后的因子(HR=2.100,95%CI=1.541~2.861,P<0.05)。经过测试集验证,本预后模型也可作为1个独立的预测预后的因子(HR=1.826,95%CI=1.282~3.425,P<0.05)(表4)。
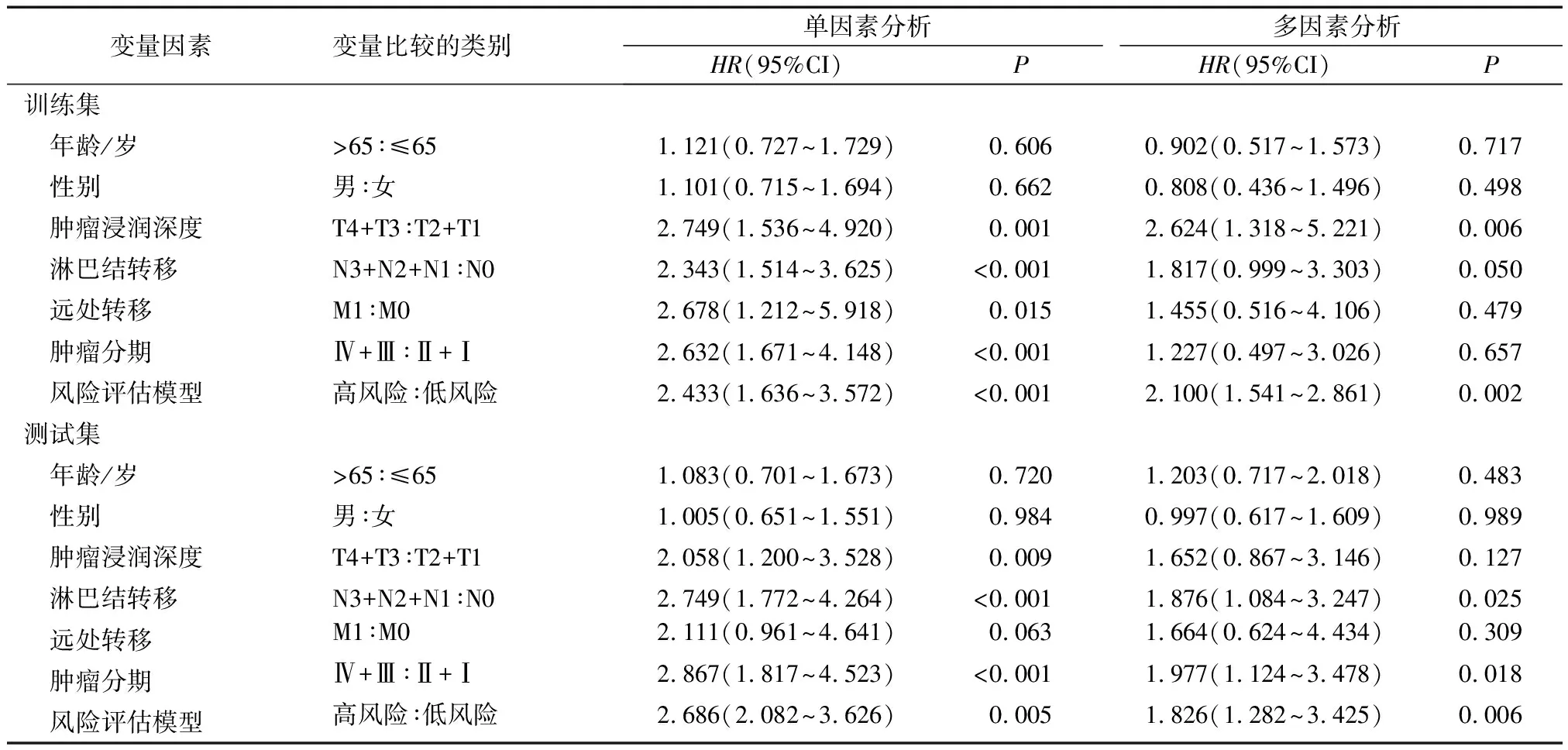
表4 肺腺癌患者训练集、测试集的单因素和多因素分析比较(n=245)
2.7 诺莫列线图建立
基于多因素Cox回归系数建立诺莫列线图(图7A);校正曲线提示,模型对3、5年的生存率均有较好的预测能力(3年的C指数为0.712,5年的C指数为0.705)(图7B)。
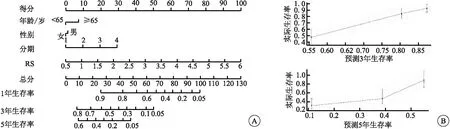
图7 模型形成诺莫列线图
2.8 基础实验验证结果
收集到的符合纳入标准的肺腺癌患者50例,其中男35例,女15例,年龄中位数为47岁,TNM分期Ⅰ期25例,Ⅱ期15例,Ⅲ期10例。实时定量PCR提示,hsa-mir-31在肺腺癌中相对量表达水平为(3.381±0.265),正常组织(0.682±0.014)(t=69.287,P=0.001),hsa-mir-1293在肺腺癌中相对表达量表达水平为(1.862±0.054),正常组织(0.161±0.021)(t=217.352,P=0.004),hsa-mir-548f-1在肺腺癌中相对表达量表达水平为(0.967±0.179),正常组织(0.217±0.041)(t=28.046,P=0.001)。3个miRNA在肺腺癌组织中表达均较高,差异有统计学意义(P<0.05)(图8)。
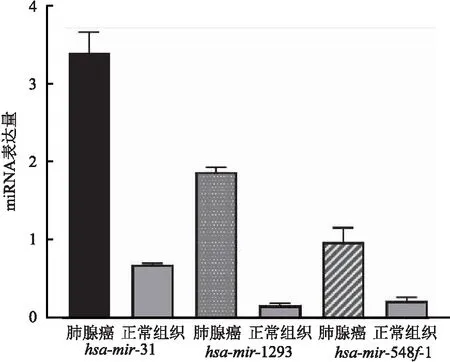
图8 3个miRNA在肺腺癌组织和癌旁正常组织的表达
3 讨论
miRNA为一种转录长度约22个核苷酸的非编码RNA,伴有非完整的特异性开放阅读框,同时缺乏编码蛋白质的功能[11]。目前许多研究[12-13]证实,miRNA能够通过组蛋白修饰、染色质异构、RNA代谢等生物学过程调控自噬基因的表达。多种肿瘤和miRNA的异常表达紧密相关,miRNA既可作为肿瘤抑制因子,又可作为促肿瘤生长因子[14]。有研究[15]报道,在非小细胞肺癌中,肿瘤抑制因子有let-7家族、miR-200、miR-486等;而另一方面,miR-31、miR-212及miR-196a具有促进肺癌细胞生长的作用[16]。miRNA以通过与靶标基因mRNA的3′端非翻译区结合为主要途径,沉默靶标mRNA或者抑制mRNA的翻译,以此调控相应蛋白的表达[17]。
miRNA的过表达、缺失或突变可通过调控肿瘤的自噬基因而对肿瘤的恶性生物行为产生驱动作用。Pishkari等[18]研究表明,在甲状腺髓样癌细胞中,过表达miR-183后LC3B表达量下降,肿瘤细胞增殖速度增快。此外,一些异常表达miRNA也与肺癌的不良预后密切相关,如程永华等[19]研究表明,miR-200b在非小细胞肺癌患者血清中表达量较健康人低,且低表达miR-200b的肺癌患者淋巴结转移的概率明显增高,OS更短。尽管目前关于miRNA在肺癌自噬中的研究已有很多,但多数是研究单个miRNA对自噬的调节作用,范围较局限,自噬相关miRNA在肺腺癌中的作用和机制以及在临床预后评估中的应用仍有待进一步探索。
本研究通过生物信息学数据库TCGA和HADb,获得了肺腺癌患者自噬相关miRNA表达谱,用Perl软件与生存相关信息匹配合并。本研究发现,在肺腺癌中的自噬基因存在异常表达,这些异常与患者的预后相关。针对自噬基因的表达改变,初步筛选出了自噬基因相关的miRNA有70个。为探寻单个自噬相关miRNA和临床预后的关系,进行了单因素Cox回归分析,筛选出12个自噬相关miRNA和临床预后密切相关。但单因素Cox回归分析每次仅纳入1个变量,可能存在过度拟合的现象,进一步使用LASSO回归分析进行降维可以降低过度拟合,最终确定了3个关键的自噬相关miRNA构建模型。根据模型对每个肺腺癌患者计算预后RS,并按照预后RS的中位数值分为高危组患者和低危组患者,在高、低危组间运用K-M生存分析,绘制ROC并计算C指数,以评估该预后RS模型的预测精度,模型结果提示,低危组患者具有明显的生存优势,3、5年总生存率的AUC值均较高,预示模型具有中等程度的预测能力。最后,将年龄、性别、病理分期等纳入多因素Cox回归分析,模型的HR=2.100,95%CI=1.541~2.861,P<0.05,说明可以作为1个独立的预后因子,并且诺莫列线图提示预后模型具有一定的临床实用价值。为了更进一步验证模型中3个miRNA的临床意义,本研究收集了50例肺腺癌患者的肿瘤标本,检测这3个自噬相关miRNA表达量,结果显示,hsa-mir-31、hsa-mir-1293、hsa-mir-548f-1的表达水平在肺腺癌组织中均较高,差异有统计学意义(P<0.05),其结果与生物信息学分析预测的趋势一致。本研究中,hsa-mir-31与cdkn2a有共表达关系,研究[20]表明,hsa-mir-31是一种人体进化中高度保守的miRNA,定位于人类染色体9q21.3上,cdkn2a并不是hsa-mir-31靶基因,不能直接受到hsa-mir-31的调控,两者共表达关系可能与hsa-mir-31与cdkn2a位置相邻有关,关于hsa-mir-1293,hsa-mir-548f-1和肺癌的研究报道较少,其与gapdh、birc5的机制有待进一步研究。
虽然本研究模型中的3个miRNA与肺腺癌预后密切相关,且该模型可以作为独立的预后因子,但是目前暂缺乏关于它们调控机制的基础实验研究,所以它们在肺腺癌中所发生的作用机制仍不清楚,有待进一步探索。另外,本研究采用的生物信息分析方法和工具较多,利用系统方法处理大量的数据是其优势,但仍然存在一定的不足之处:1)大部分数据均来自于TCGA数据库,未通过其他数据库再次对其验证;2)预后RS模型仅纳入了自噬相关miRNA表达水平,未考虑其他基因改变,如LncRNA、circRNA等表达水平改变对预后的影响。在下一步研究中,将结合本研究的验证数据和随访信息,开展更为深入的生物学水平机制研究,同时考虑纳入更多可能影响临床预后的因素,如吸烟情况、LncRNA、circRNA表达水平等,以期构建更稳定和可靠的预后RS模型服务于肺癌患者。
综上所述,本研究通过挖掘TCGA数据库,构建了1个基于3个自噬相关miRNA的肺腺癌预后模型,预测准确性中等,且该模型可以作为独立的预后因子,可能为肺腺癌机制的研究提供一定理论依据,为探索肺腺癌相关分子标志物及个体化治疗方案提供一定参考。
猜你喜欢
延安大学学报(医学科学版)(2022年2期)2022-11-23
临床军医杂志(2022年7期)2022-08-06
昆明医科大学学报(2022年2期)2022-03-29
中国药学药品知识仓库(2022年1期)2022-03-23
昆明医科大学学报(2022年1期)2022-02-28
祝您健康·文摘版(2021年7期)2021-07-17
养生阅刊(2020年7期)2020-08-16
祝您健康·文摘版(2020年7期)2020-07-13
饮食与健康·下旬刊(2019年10期)2019-03-09
世界知识(2018年19期)2018-11-21