
特发性肺纤维化相关信号通路及治疗药物研究进展
2022-08-16 08:43来志龙徐寒梅赵万洲胡加亮
药学进展 2022年7期
来志龙,徐寒梅,赵万洲,胡加亮*
(1. 中国药科大学,江苏 南京 211198;2. 南京安吉生物科技有限公司,江苏 南京 210046)
特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)是特发性间质肺炎的最常见形式之一,是一种慢性的进行性肺部疾病,其特征是肺结构和功能不可逆转。IPF死亡率很高,自诊断之日起中位生存期为2 ~ 3年,呼吸衰竭是最常见的死亡原因。IPF的纤维化过程最初被认为由慢性炎症导致,随着研究的深入,对其发病机制的理解已经演变成遗传学、环境因素和肺泡上皮细胞慢性微损伤共同作用导致肺部修复过程不良进而发展为纤维化[1]。一些外源性因素(感染、毒素、烟雾)或内源性因素(炎症、氧化应激、异常的免疫反应)引起肺泡上皮损伤,会触发转化生长因子(TGF)-β、肿瘤坏死因子(TNF)-α等纤维化细胞因子和结缔组织生长因子(CTGF)、胰岛素样生长因子(IGF)-1和血小板衍生生长因子(PDGF)等生长因子的释放。纤维化细胞因子与生长因子在局部和循环水平的升高会在一定程度上刺激肺成纤维细胞的活化和增殖。活化的肺成纤维细胞分化为表达α-平滑肌肌动蛋白(α-SMA)的肺肌成纤维细胞后[2],产生过量的纤维化肺组织的细胞外基质(ECM)蛋白[3]。肺肌成纤维细胞还通过调节基质金属蛋白酶(MMP)及其抑制剂[MMP组织抑制剂(TIMP)]之间的平衡来改善ECM的代谢,从而促进IPF的进程[4]。成纤维细胞的活化、肌成纤维细胞的增殖以及ECM的过度累积在各种原因导致的纤维化形成过程中都发挥关键作用。多条信号通路调节IPF的发生发展,包括TGF-β/Smad信号通路、Wnt/β-catenin信号通路、血管内皮生长因子(VEGF)信号通路、成纤维细胞因子(FGF)信号通路、PDGF信号通路[5]以及磷酸肌醇-3-激酶(PI3K)-蛋白激酶B(Akt)信号通路等[6]。这些信号通路被激活后将调节信号传递至细胞,从而调控细胞的增殖、凋亡、分化/转分化、迁移等生物学行为。靶向信号通路中的关键蛋白可以延缓IPF的发展,延长IPF患者的生存期。因此,信号通路的研究在IPF病理学发展中至关重要。随着对IPF的深入研究,人们对IPF的发病机制也更加了解,从而为IPF诊断和治疗提供了新的途径和方法。本文对IPF研究进展进行综述,介绍IPF相关信号通路及其临床在研药物最新进展。
1 特发性肺纤维化相关信号通路
1.1 TGF-β/Smad信号通路
TGF-β是转化生长因子超家族的多功能细胞因子,自1981年被发现其具有诱导成纤维细胞转分化的能力以来,已发现属于该超家族的成员超过40个。在哺乳动物体内,这些细胞因子发挥调节细胞生长、分化、迁移、凋亡,以及细胞外基质(ECM)的产生等作用[4]。TGF-β具有3种同工型,包括TGF-β1、TGF-β2和TGF-β3[7]。多种纤维化疾病的发生发展过程会激活这3种转化生长因子[8]。TGF-β1是纤维化和炎症发展的关键介质,TGF-β1的2个受体TGF-βRⅠ和TGF-βRⅡ在上皮-间质转化(EMT)和纤维化形成过程中起关键作用[8]。其下游分子包括Smad2、Smad3、Smad4和Smad7参与TGF-β1诱导的EMT,而Smad7阻断Smad3的表达[8]。TGF-β1会激活包括Smad2和Smad3在内的下游介体,但受到Smad7表达的负调控。因此在纤维化病理条件下,Smad2和Smad3表达均被上调,而Smad7被下调。
TGF-β1首先以无活性的形式分泌到ECM中。TGF-β1与潜伏期相关肽(LAP)形成复合物从而储存TGF-β1[9]。这一复合物还有另外一种成分,该成分是潜在的TGF-β结合蛋白,可促进ECM中TGF-β1的储存。TGF-β1被细胞中的各种信号激活后会转变为通过二硫键连接的二聚体。TGF-β1二聚体与TGF-βRⅡ结合,使TGF-βRⅡ磷酸化,磷酸化的TGF-βRⅡ激活TGF-βRⅠ激酶。激活的TGFβRⅠ磷酸化细胞质介体Smad2和/或Smad3,并与Smad4形成异源三聚体复合物,该复合物易位进入细胞核,结合共有序列,直接或间接调节基因转录[8]。该信号传导过程触发了一系列细胞内信号,最终刺激核转录因子Snail和Twist的转录,抑制内皮标志物的产生并激活间充质标志物的表达。TGF-β的激活会导致过多的ECM成分产生,包括原纤维蛋白、纤连蛋白等,它们直接或间接控制TGF-β的活化[10]。
TGF-β/Smad是一种多效性信号通路,在炎症、伤口愈合和纤维化过程中起关键作用,同时也是组织纤维化中ECM沉积和EMT过程的重要诱因[11]。TGF-β1通过Smad7的过表达抑制Smad3/4作用[12],Smad7可通过抑制Smurf2泛素连接蛋白的泛素化来抑制Smad2和Smad3的磷酸化,从而起到抑制剂的作用。此过程有助于TGF-βRⅠ和RⅡ复合物的降解,最终结果是抑制TGF-β1的信号传导。TGF-β诱导人肺泡上皮细胞中的C/EBPβ乙酰化从而增加α-SMA表达[2]。此外TGF-β通过广泛的分子相互作用网络促进EndMT或其他信号通路(包括Wnt和Notch)相互作用来发挥作用[13]。HSP90是热休克蛋白中的一种[14],其在IPF患者中增加[15],近来被认为是肺纤维化的生物标志物和潜在的治疗靶点。特别是IPF 患者中的 HSP90 激活后会导致细胞外基质蛋白和胶原蛋白合成增加和异常沉积。最近研究表明,HSP90 的抑制会导致TGF-β/Smad信号通路的阻断[16-17]。IPF患者气道上皮和成纤维细胞中的TGF-β1升高会引起成纤维细胞分化和异常的损伤反应,从而加剧IPF的进行性纤维化[18]。研究表明,在小鼠模型中,抑制TGF-β信号可延迟肺纤维化的进展[19]。肺泡巨噬细胞是TGF-β1的重要来源,M2巨噬细胞与小鼠肺上皮细胞共培养可诱导上皮细胞发生EMT。TGF-β受体抑制剂LY2109761预处理巨噬细胞可阻断EMT,表明肺泡M2巨噬细胞可通过TGF-β1/Smad信号通路诱导EMT的发生[20]。在过表达TGF-β1的转基因小鼠模型中,通过限制肺泡巨噬细胞的蓄积,可抑制博来霉素诱导的肺纤维化[21]。尽管靶向TGF-β信号通路中的一些分子的治疗方法可减轻模型鼠的肺纤维化[11,22],但这些研究仍停留在实验室阶段,尚未进入临床研究。TGF-β对一些正常的生理功能的执行亦必不可少,抑制TGF-β活性可能导致异常的免疫活化、上皮增生和伤口愈合受损。因此,在不干扰TGF-β稳态功能的情况下,将介导纤维化的TGF-β下游信号传导效应子作为靶点,或可为纤维化的治疗带来新思路。
1.2 Wnt/β-catenin信号通路
Wnt包括19种分泌型糖蛋白,其主要功能是调节哺乳动物胚胎发育和机体的损伤[23]。Wnt信号通路主要包括经典Wnt/β-catenin通路、非经典平面细胞极性通路和非经典Wnt/钙通路。当Wnt与共受体低密度脂蛋白相关蛋白(LRP)结合到细胞表面的Frizzled受体时,就会启动Wnt/β-catenin通路,导致Dishevelled(DSH)受体家族蛋白质的激活,最终使细胞核内β-catenin的水平发生变化。DSH是细胞膜相关Wnt受体复合物的关键成分,它与Wnt结合后被激活,并抑制下游蛋白质复合物,包括axin、GSK-3、APC蛋白[24]。Axin/GSK-3/APC复合体可促进细胞内信号分子β-catenin的降解。当β-catenin降解复合物被抑制后,胞浆内的β-catenin得以稳定存在,部分β-catenin进入细胞核与TCF/LEF转录因子家族作用并促进特定基因的表达,包括MMP基因、细胞周期调节因子基因及致癌基因。在人类的肺组织中,多种Wnt蛋白被检测到,包 括Wnt2、Wnt3A、Wnt5A、Wnt5B、Wnt7B、Wnt10A、Wnt11和Wnt13[23]。
Wnt/β-catenin信号传导在肺部许多病理过程中起重要作用,例如炎症、重塑和纤维化[25]。IPF患者的肺活检结果表明,经典Wnt/β-catenin信号通路的高度激活与组织修复和成纤维细胞激活有关。有研究也表明Wnt/β-catenin信号传导参与纤维化发展过程中EMT的诱导[25]。Wnt1、Wnt3A、Wnt7B、Wnt10B、Fzd2、Fzd3和 β-catenin表达在IPF患者肺组织中显著增加[26]。据报道,Wnt5A和Wnt5B配体能通过TGF-β对肺成纤维细胞分化产生影响[27]。许多研究将Wnt信号通路置于TGF-β信号的下游。Wnt/β-catenin信号通路水平的提高会促进成纤维细胞的迁移和增殖,这表明Wnt/β-catenin的激活可能是肺纤维化的常见特征。然而也有文献报道Wnt信号可能作用于TGF-β的上游[28]。Wnt信号传导上调可能导致上皮细胞表达过量TGF-β,并诱导EMT来调节组织修复和诱导ECM(如胶原蛋白、MMP)的累积。Wnt信号异常暗示着IPF的发生和发展,导致EMT,以及ECM沉积、肺成纤维细胞增殖和肌成纤维细胞分化[29]。Wnt信号通路的下调抑制了肌成纤维细胞分化,从而改善了肺纤维化病变[30]。IPF动物模型中,Wnt/β-catenin信号通路被明显激活[31]。Wnt/β-catenin通路的阻滞可以减弱小鼠肺纤维化[29]。Wnt/β-catenin信号通路还能与IPF进展中其他信号通路相互串扰[32]。
1.3 VEGF、FGF、PDGF信号通路
VEGF家 族 主要 包括VEGF-A、VEGF-B、VEGF-C和VEGF-D。VEGF发挥生物学作用依赖VEGF与酪氨酸激酶受体VEGFR1和VEGFR2的结合。VEGFR2是肺中细胞VEGF信号的主要传导者,当游离的VEGF与细胞表面的VEGFR结合后,会导致VEGFR发生磷酸化并激活下游信号通路,主要包括PI3K-Akt信号通路和黏着斑激酶(FAK)途径,这是导致纤维化发展的原因[33]。
活化的FAK通过与TGF-β整合促进细胞迁移、浸润并抵抗细胞凋亡[34]。抑制FAK可以预防实验性肺纤维化和肺肌成纤维细胞的形成[35]。在博来霉素诱导的IPF模型中,FAK抑制剂或siRNA介导的FAK沉默可显著终止肺纤维化[36]。在注射博来霉素的动物的肺组织中,VEGF的表达显著增加[37],使用VEGFR拮抗剂后可以减轻小鼠肺纤维化组织的病理性纤维化和胶原沉积[38]。VEGF-A还显示出刺激PDGF受体(PDGFR)的活性,从而调节间充质细胞的迁移和增殖。在IPF患者肺中,VEGF主要由Ⅱ型肺泡上皮细胞和肺成纤维细胞分泌。
FGF家族包括20多种分泌蛋白,FGF通过与细胞表面具有酪氨酸激酶活性的单程跨膜蛋白FGFR结合,调控下游的一些信号通路,例如Ras/MAPK/ERK-1、PI3K/Akt等,进而调控细胞的增殖、活化和迁移[39]。FGF家族的某些成员通过促进成纤维细胞的促有丝分裂活性而发挥促纤维化作用[40]。一些FGF通过促进上皮细胞的再生和增殖发挥抗纤维化作用。FGF-1抑制TGF-β1刺激的成肌纤维细胞分化和EMT发挥抗纤维化作用[41]。FGF-10维持肺泡上皮祖细胞的克隆扩增和分化,并保护它们免受氧化应激、石棉诱导的DNA损伤和细胞凋亡[42-43]。与稳定的IPF相比,进行性IPF患者的肺泡间充质基质细胞中FGF-10表达显著降低[44-45]。FGF-9和FGF-18促进人肺成纤维细胞的存活和迁移,并在体外抑制肌成纤维细胞的分化[46]。FGF-18降低肌成纤维细胞的分化,而FGF-7和FGF-10刺激肺泡上皮细胞的增殖并减少上皮损伤和凋亡[43]。碱性成纤维细胞生长因子(bFGF)的FGF-2 是成纤维细胞、气道平滑肌细胞和Ⅱ型肺泡上皮细胞的有效有丝分裂原[47],并诱导肺成纤维细胞和肌成纤维细胞中的胶原蛋白合成[48]。TGF-β信号上调FGF-2的表达,FGF-2通过激活MAPK和PI3K-Akt途径协同增强TGF-β1诱导的成纤维细胞增殖[48]。靶向间质FGF信号传导可以作为治疗IPF的策略。
PDGF家 族 由PDGF-A、PDGF-B、PDGF-C和PDGF-D之间以二硫键结合形成的同二聚体或异二聚体组成,它们以5种亚型的形式存在,包括AA、AB、BB、CC和 DD。PDGF二聚体以不同的亲和力结合并磷酸化2个受体酪氨酸激酶(PDGFR-α和PDGFR-β)的同二聚体或异二聚体。PDGFR-α和PDGFR-β往往来自器官中的间充质细胞。PDGF与PDGFR-α或PDGFR-β结合后,磷酸化受体激活下游信号转导途径,包括MAPK、PI3K/Akt等[49]。PDGF 是一种常见的生长因子,可在许多细胞中产生,包括巨噬细胞、血小板、内皮细胞和成纤维细胞[50]。PDGF是一种有效的成纤维细胞有丝分裂原,并在肌成纤维细胞的增殖中发挥重要作用[51]。PDGF 的过量产生可诱发心脏、肝脏和肾脏纤维化。PDGF与小鼠骨髓、肺、肾、肝和心脏的纤维化有关。PDGF通过其线粒体特性和趋化特性促进纤维化[52]。PDGF还可以通过串扰机制与TGF-β协同作用以增强纤维化,其中包括PDGF对TGF-β水平的调节[49]。在IPF患者的肺组织中,上皮细胞和肺泡巨噬细胞中PDGF的表达增加[53]。用PDGF处理人肺成纤维细胞可通过激活MAPK途径促进胶原Ⅰ和α-SMA的表达[54]。在IPF的动物模型中,靶向PDGFR-β可以缓解博来霉素诱导的IPF[55]。因此,PDGF被认为是参与肺纤维化的重要致病因素。
尼达尼布是少数临床上获批的抗纤维化药物之一,是一种酪氨酸激酶抑制剂,可阻断PDGFR以及FGFR和VEGFR的活性。在IPF患者中,尼达尼布可抑制成纤维细胞向肌成纤维细胞的分化和肌成纤维的增殖。另一方面,伊马替尼也可抑制PDGFR酪氨酸激酶,但在IPF患者的临床试验中未显示出疗效。 因此,仍不确定尼达尼布是否主要通过抑制PDGFR酪氨酸激酶发挥抗纤维化疗效,以及PDGFR是否为纤维化的主要靶标。
1.4 PI3K-Akt信号通路
PI3K-Akt信号通路在细胞生长、增殖和凋亡中起着重要作用。PI3K催化磷脂酰肌醇二磷酸(PIP2)的3位羟基磷酸化,生成磷脂酰肌醇三磷酸(PIP3),激活Akt;活化的Akt可以通过信号分子调节细胞内信号传导并保护细胞免于凋亡[56]。Akt的激活往往会导致疾病的恶化和预后不良,在IPF患者的肺成纤维细胞中,PI3K-Akt信号被异常激活,磷脂酰肌醇-3,4,5-三磷酸-3-磷酸酶(PTEN)的活性较低[57]。在这种情况下,PI3K-Akt信号通路中Akt的靶基因FoxO3a的功能被抑制。FoxO3a是细胞周期的强效抑制剂之一,同时也是凋亡的促进剂[58]。FoxO3a表达过低可导致肺肌成纤维细胞免于凋亡[59]。在正常的成纤维细胞中,PTEN的激活会抑制PI3K-Akt信号,而成纤维细胞被促进凋亡,最终抑制成纤维细胞的转化和蓄积[60]。因此,PI3K-Akt信号通路是IPF必不可少的治疗靶标。
在IPF的调节过程中,PI3K-Akt信号通路与其他信号通路有许多相互作用。例如,PI3K可被酪氨酸蛋白激酶(Src)激活,Src可被VEGF信号通路中的VEGFR激活[61]。FAK和PI3K之间的信号传递可以调节细胞存活、细胞凋亡和细胞周期[62]。整合素蛋白是重要的细胞表面受体,它们调节细胞基质与黏着斑途径之间的联系。其中整合素β1(ITGB1)在成纤维细胞中过表达,影响成纤维细胞与纤连蛋白和胶原蛋白的黏附。整合素和ECM蛋白之间的双向调节将影响成纤维细胞的分化以及TGF-β1和FAK的激活[63]。CXCL16/CXCR6通过激活PI3KAKT-FOXO3a信号通路,增加人肺成纤维细胞的增殖、迁移和胶原合成,从而加速纤维化[64]。此外,在EMT期间可以检测到PI3K-Akt信号增强,并且伴随着Snail1的表达增多和E-钙黏蛋白的抑制[65],这些通路之间的串扰和相互作用形成了复杂的通路网络。
2 特发性肺纤维化治疗药物
IPF是特发性间质性肺炎的最常见类型,其发病频率与胃癌、脑癌和睾丸癌类似。近年来,特发性肺纤维化的发病率呈上升趋势。特发性肺纤维化在男性中更为常见,在50岁以下的人群中很少见(诊断中位年龄约为65岁),自诊断之日起中位生存期为3 ~ 4年[66]。目前针对IPF 的上市药物主要有吡非尼酮和尼达尼布。吡非尼酮于2008年在日本获批准上市,口服吡非尼酮可减缓疾病进展和延长IPF 患者寿命。其主要是通过下调促纤维化生长因子TGF-β1表达发挥抗纤维化作用。尼达尼布是三重酪氨酸激酶抑制剂,其可通过抑制VEGFR、PDGFR以及FGFR 的信号通路来治疗IPF。随着对IPF信号通路和发病机制的深入探索,越来越多的靶点被用于抗纤维化的研究。目前处于Ⅱ期和Ⅲ期临床研究阶段的IPF治疗药物有10余种。
2.1 Pentraxin-2重组蛋白
PRM-151是由罗氏研发的一种Pentraxin-2(PTX-2)的重组蛋白。一项随机、双盲的Ⅱ期临床试验(NCT02550873)评估了 PRM-151在IPF患者中的疗效和安全性。结果显示,IPF患者的用力肺活量(forced vital capacity,FVC)的下降程度具有统计学意义,达到了主要终点;在次要终点中,只有患者的 6 min步行距离的变化具有统计学意义[67]。这些结果保证了Ⅲ期随机试验 (NCT04552899)的顺利开展。目前Ⅲ期随机试验正在进行患者招募,该试验旨在比较PRM-151与安慰剂在IPF患者中的疗效、安全性和药代动力学(PK)。
2.2 结缔组织生长因子抗体
Pamrevlumab(FG-3019)是 由Fibrogen研 发 的一种结缔组织生长因子(CTGF)单克隆抗体。一项开标记Ⅱa期研究(NCT01262001)评估了pamrevlumab在IPF患者中的安全性、耐受性并提供了有关疗效的初步数据。结果显示各组患者仅有轻微不良反应,pamrevlumab组中1名患者表现出肺部的网状纤维化减少[68]。随后又进行了后续的Ⅱb期安慰剂对照试验(NCT01890265),结果显示,pamrevlumab组与安慰剂组相比,IPF患者的FVC下降幅度降低,IPF疾病进展更加缓慢[69]。基于上述结果,pamrevlumab的Ⅲ期临床研究(NCT03955146)进入患者招募阶段,该研究旨在评估pamrevlumab在IPF患者中的疗效和安全性。
2.3 IL-13和IL-4抗体
Lebrikizumab是由罗氏研发的一种可溶性IL-13的人源化单克隆抗体。一项Ⅱ期临床研究(NCT01872689)对lebrikizumab作用进行了评估。结果表明:在主要终点评价中,lebrikizumab作为单一疗法或吡非尼酮补充疗法在减缓IPF患者的肺功能下降方面是无效的;但在次要终点评价中,lebrikizumab和吡非尼酮联合治疗降低了IPF患者的死亡率,并延长了首次急性IPF患者的疾病恶化或死亡时间。
SAR156597是由赛诺菲研发的一种靶向IL-4和IL-13的人源化单克隆抗体。一项Ⅱ期临床研究(NCT02345070)已完成,但结果未能证明SAR15697对IPF患者具有治疗作用。
2.4 脱脂转化酶途径抑制剂
BMS-986278是一种新型脱脂转化酶(LPA)1拮抗剂,具有较高口服生物利用度和抗纤维化活性,其在健康受试者中进行的单/多剂量递增Ⅰ期研究(NCT03981094)已完成。目前一项多中心、随机的Ⅱ期临床试验(NCT0430868)已开始招募IPF患者,该试验主要终点为患者的FVC下降速率。
BMS-986020是一种口服选择性LPA1受体拮抗剂,已被美国FDA授予孤儿药资格,用于治疗IPF。一项Ⅱ期临床研究(NCT01766817)表明,BMS-986020与安慰剂相比可显著减缓IPF患者FVC下降的速度[70]。
2.5 细胞内激酶抑制剂
CC-90001是由Celgene研发的c-Jun氨基端激酶1(JNK1)抑制剂。一项随机、双盲、Ⅱb期研究(NCT03142191)对CC-90001进行了评估,主要终点为IPF患者的FVC下降速率。该项研究结果目前暂未公布。
2.6 Galectin-3抑制剂
TD139是由Galecto Biotech研发的Galectin-3抑制剂。一项随机、双盲、安慰剂对照的Ⅱb期研究(NCT03832946)正在进行患者招募,该研究的目的是评价TD139的有效性和安全性。主要终点是IPF患者的FVC下降速率,次要终点包括疾病进展情况、死亡时间或首次住院时间。
2.7 趋化因子2抑制剂
CNTO-888是强生研发的一种对趋化因子(CCL)2具有高亲和力和特异性的人单克隆抗体。一项Ⅱ期临床研究(NCT00786201)对CNTO-888在IPF患者中的安全性和有效性进行了评价。然而结果显示,CNTO-888组与安慰剂组相比,IPF患者的FVC下降程度更大,且游离CCL2水平在24周和52周均高于基线[71],提示CNTO-888并未改善IPF患者的肺功能。
2.8 整合素抑制剂
BG00011(STX-100)是一种人源化单克隆抗体,通过针对αvβ6整合素阻断TGF-β通路。一项随机、双盲、安慰剂对照的Ⅱ期临床研究(NCT01371305)评估了BG00011的安全性和耐受性,目前试验结果尚未公开。
PLN-74809是由斯坦福大学研发的αvβ6和αvβ1整合素的选择性双重抑制剂,一项随机、双盲、Ⅱa期临床研究(NCT04396756)正在进行患者招募,该研究旨在评估PLN-74809在IPF患者中的安全性、耐受性和药代动力学。研究终点包括在治疗过程中的相关不良事件、IPF患者FVC下降速率和HRCT定量纤维化评分。此外另一项Ⅱa期研究(NCT04072315)将通过PET扫描测定IPF患者在口服PLN-74809期间肺泡上皮细胞上αvβ6受体占位的变化,以确定靶标参与率。
2.9 NADPH氧化酶-1/-4抑制剂
GKT137831是一种新型NADPH氧化酶(NOX)-1/-4双重抑制剂,由阿拉巴马大学通过高通量筛选开发。一项随机、安慰剂对照的Ⅱ期临床研究(NCT03865927)将对GKT137831在IPF患者中的有效性进行评价,该研究正在进行患者招募。
2.10 B细胞激活因子受体抑制剂
VAY736是诺华制药研发的一种针对B细胞激活因子受体(BAFF-R)的单克隆抗体。一项随机、安慰剂对照、多中心的Ⅱ期临床研究(NCT03287414)目前正在招募IPF患者,评估VAY736的疗效、安全性和耐受性。该研究主要终点是患者在治疗48周后,患者的FVC较治疗前的变化,次要终点包括无进展生存期和运动耐受性等。
2.11 RNA药物
ND-L02-s0201是百时美施贵宝研发的一种脂质纳米粒,其包裹的siRNA可阻止热休克蛋白47(HSP47)的翻译。一项随机的Ⅱ期临床研究(NCT03538301)正在招募轻至中度功能损害的IPF患者,旨在评估ND-L02-s0201的安全性、耐受性、生物活性和药代动力学。该研究的主要终点是与治疗相关的不良事件的患者数量。
3 结语
本文对在研的IPF药物、相关靶点和作用机制进行了简要总结(见表1)。
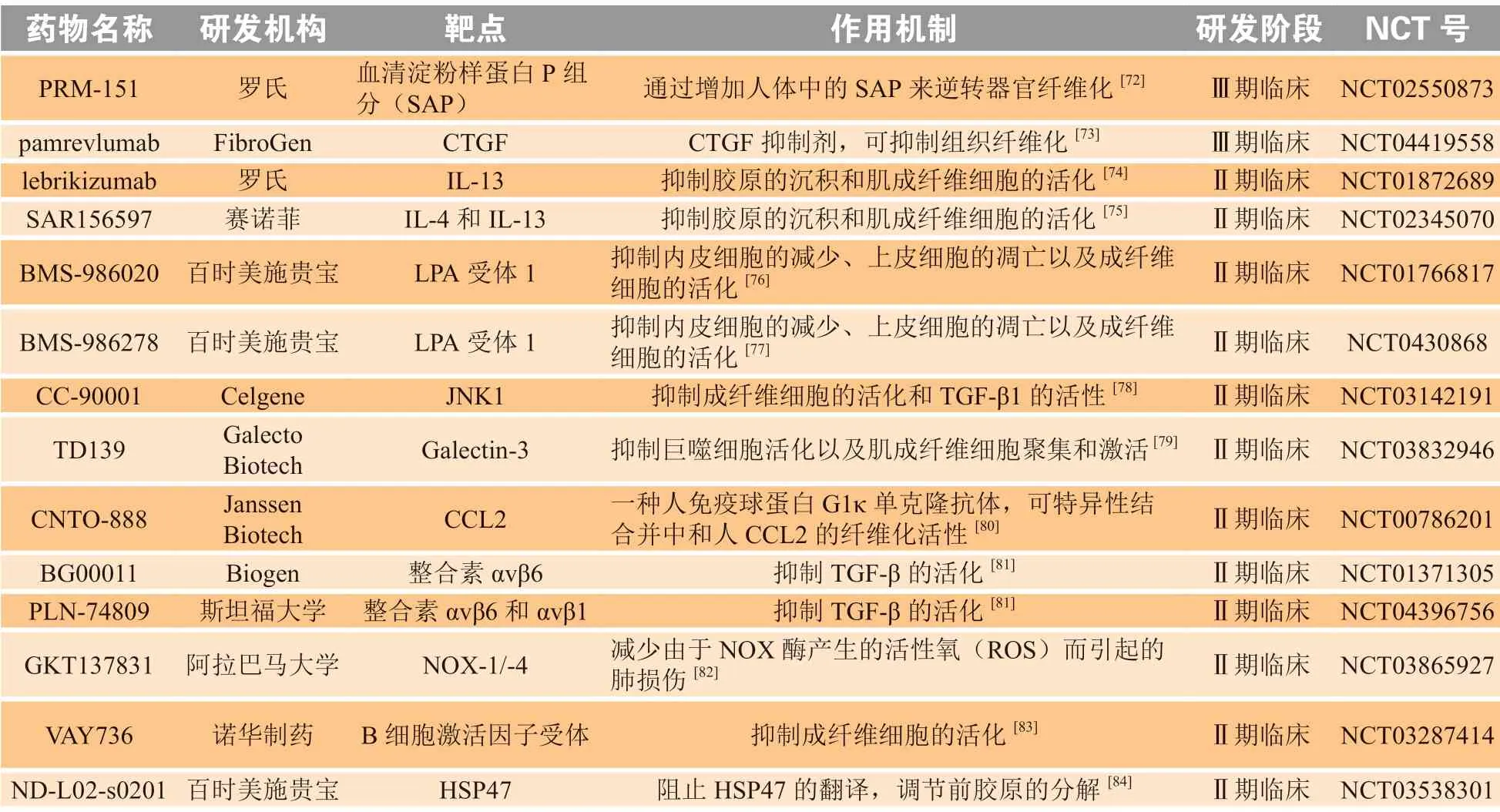
表1 临床在研的特发性肺纤维化治疗药物Table 1 Drugs in clinical development for the treatment of idiopathic pulmonary fibrosis
IPF是一种复杂的疾病,在其发生发展过程中,许多细胞因子和信号传导途径异常。尽管本文概述了多条IPF相关信号通路,但还有许多其他信号通路[例如TNF-α信号通路、Janus激酶/信号通路、转录激活因子(JAK-STAT)信号通路和哺乳动物雷帕霉素靶蛋白(mTOR)信号通路等]以及更多在IPF病理过程中起着重要作用的蛋白仍在探索中。所有这些信号传导途径都不能单独起作用,客观上存在着一定的串扰和相互作用,并且共同调节IPF的发展。随着医学发展和生物学数据的积累,人们对IPF相关的信号传导途径的认知也日趋全面。通过对IPF信号通路展开深入研究,可更好地探索与疾病相关的基因及其功能,以及发掘治疗靶点和可 用于早期诊断的新型生物标志物。
猜你喜欢
中老年保健(2022年2期)2022-11-25
中国心脏起搏与心电生理杂志(2022年3期)2022-11-23
昆明医科大学学报(2022年4期)2022-05-23
家庭医学(2022年5期)2022-04-27
井冈山大学学报(自然科学版)(2022年1期)2022-02-28
昆明医科大学报(2019年3期)2019-10-21
科学24小时(2018年1期)2018-01-10
农家科技下旬刊(2016年8期)2017-05-05
现代养生·下半月(2016年6期)2016-10-21
中国民族民间医药·下半月(2011年10期)2011-12-27
