
NLRP3炎症小体在动脉粥样硬化中的作用机制及相关治疗策略*
2022-08-15 01:43潘亚慕
华中科技大学学报(医学版) 2022年3期
刘 瑶,潘亚慕,付 坚△,金 莹△
十堰市人民医院 1神经疾病中心 2心血管内科临床研究所,十堰 442000
动脉粥样硬化(atherosclerosis,AS)是冠心病及卒中等心脑血管事件发生的重要病理基础,它是由内皮功能障碍和内皮下脂蛋白积聚相互作用引起的一种慢性适应不良,且不可逆转的炎症性疾病[1]。NLRP3炎症小体参与机体多种危险因素驱动的血管炎症反应,对动脉粥样硬化的发生及发展极其重要。本文主要概括了脂质及体细胞TET2突变等危险因素介导NLRP3炎症小体的激活作用及机制,并简述NLRP3炎症小体靶向治疗的现状,为探讨冠状动脉粥样硬化性心脏病(冠心病,coronary heart disease,CHD)及卒中等疾病的治疗提供新思路。
1 NLRP3炎症小体及其激活过程
NLRP3炎症小体主要由受体蛋白NLRP3、接头蛋白ASC和效应蛋白pro-Caspase-1构成[2]。NEK7作为NLRP3炎症小体的新成员,可直接结合NLRP3并调控其蛋白复合物的多聚化和激活[3]。机体损伤相关分子模式(damage-associated molecular patterns,DAMPs)或病原相关分子模式(pathogen-associated molecular patterns,PAMPs)促进NLRP3炎症小体的组装与活化。首先,启动信号(priming signal)刺激NF-κB信号通路诱导IκB磷酸化[4],促进NLRP3和IL-1β等前体蛋白转录上调[5];其次,NLRP3炎症小体响应激活信号(activation signal)招募并组装ASC和pro-Caspase-1并促使后者酶解为Caspase-1,活化的Caspase-1加工修饰IL-1β及IL-18等促炎因子并切割Gasdermin-D(GSDMD)[6],GSDMD-N端与细胞膜结合形成孔隙促进炎性因子释放[7-8],诱发细胞焦亡等一系列生物学效应[9],具体激活途径见图1。此外,Caspase-8或Caspase-9依赖性细胞凋亡通路中,糖蛋白Pannexin-1亦可促进NLRP3炎症小体活化,从而介导细胞死亡[10]。NLRP3炎症小体异常激活造成宿主损伤,与炎症性疾病、自身免疫性疾病、代谢性疾病及肿瘤等疾病的发生和发展有密切关系。
2 NLRP3炎症小体活化的影响因素
有关NLRP3炎症小体的激活机制尚未完全阐明,其活化的影响因素可以概括为以下几种。①泛素化状态:NLRP3被K48和K63泛素链多聚泛素化来维持其失活状态。泛素特异性肽酶1-分配因子1(ubiquitin specific peptidase 1-associated factor 1,UAF1)通过靶向NLRP3和p65(NF-κB)增强NLRP3和pro-IL-1β的表达[11]。ABRO1募集BRCC3(Lys63-特异性去泛素化酶)并去除K63连接的NLRP3泛素化[12],促进NLRP3炎症小体的组装和激活[13]。也有研究提示NLRP3泛素化促进NLRP3炎症小体活化。调控NLRP3泛素化被认为是NLRP3炎症小体激活的关键步骤[14]。②细胞器功能:线粒体稳态失衡导致NAD(+)依赖性的α-微管蛋白脱乙酰酶失活,乙酰化的α-微管蛋白介导线粒体动力蛋白依赖性运输,促进线粒体ASC与内质网NLRP3结合及组装[15]。动力蛋白适配器组蛋白脱乙酰基酶6(the dynein adapter histone deacetylase 6,HADC6)介导炎症小体的微管组织中心(the microtubule-organizing center,MTOC)定位和活化[16],MTOC充当枢纽促进自噬体和溶酶体融合。溶酶体释放组织蛋白酶诱导氧化应激,导致大量活性氧(ROS)产生及硫氧还蛋白相互作用蛋白/硫氧还蛋白-1(TXNIP/TRX-1)分离,在内质网(ER)水平与NLRP3相互作用,促进NLRP3炎症小体激活及促炎因子IL-1β释放[17]。此外,机体接受刺激诱导反式高尔基体网络分解为分散的反式高尔基体网络(dTGN),后者充当炎症体复合物聚集的支架促使NLRP3炎症小体组装和激活[18]。③离子通道开放:ATP结合嘌呤能受体(P2X7R)激活诱导钾外流、钙内流,导致胞内钙离子持续增加[19];TWIK2亦作为巨噬细胞中钾流出通道[20],导致钾外流、线粒体损伤和ROS产生,氯化物细胞通道(CLICs)作用于钾外排-线粒体ROS轴的下游[21],促进NEK7-NLRP3相互作用和炎症小体信号转导[22]。最新研究显示[23],特征性神经损伤诱导蛋白-1(nerve injury-induced protein 1,NINJ1)介导质膜破裂和损伤相关分子模式释放,启动并扩大炎症反应,参与细胞焦亡、凋亡和坏死等过程。以上激活及调控机制仅在部分途径实现,具有一定局限性,其关键激活机制仍有待进一步研究。
3 NLRP3炎症小体在动脉粥样硬化发生发展中的作用
NLRP3炎症小体是脂质驱动血管炎症反应的关键因素,在动脉粥样硬化的发生和发展中起着根本性的作用[24]。研究显示,NLRP3炎症小体在人颈动脉粥样硬化斑块中表达增加,特别是在不稳定斑块中,导致动脉粥样硬化形成和斑块易损性增加[25]。糖尿病性动脉粥样硬化小鼠模型中,NLRP3敲低可抑制NLRP3炎症小体激活及内膜中粘附分子ICAM-1和VCAM-1的表达,减少动脉粥样硬化并稳定动脉粥样硬化斑块[26]。NLRP3炎症小体主要表达于斑块内的泡沫细胞中,可诱导IL-1β及IL-18在炎症区域迅速聚集,协同增加其生物学效应,参与血管内皮损伤及动脉粥样硬化形成[27]。总之,NLRP3炎症小体参与动脉粥样硬化斑块的形成,与斑块不稳定性密切相关,是冠状动脉粥样硬化严重程度的预测因子。
4 脂质与NLRP3炎症小体
NLRP3炎症小体将脂质及脂蛋白的动脉沉积与介导动脉粥样硬化发生发展的炎症反应联系在一起[28]。单核细胞在内皮下分化而成的巨噬细胞吞噬氧化修饰低密度脂蛋白(oxidized low density lipoprotein,oxLDL)并激活NLRP3炎症小体,导致泡沫细胞形成及胆固醇结晶体积累,NLRP3炎症小体在动脉粥样硬化病变早期即发挥重要作用[29]。oxLDL诱导非选择性阳离子通道P2X7R表达上调并增强蛋白激酶R(PKR)磷酸化,协助组装并激活NLRP3炎症小体,从而刺激巨噬细胞产生并释放炎症因子IL-1β[30]。oxLDL与脂肪转位酶CD36结合介导微血管内皮细胞的炎症反应,影响ROS生成、钾流出和组织蛋白酶B活性,促进NLRP3炎症小体激活和炎症因子的成熟分泌,参与泡沫细胞形成和动脉粥样硬化发展[31]。oxLDL在NLRP3炎症小体启动和激活过程中具有双重作用,成为动脉粥样硬化发展的主要驱动力。
动脉粥样硬化发展的最初病理改变是内皮损伤,胆固醇通过低密度脂蛋白(LDL)颗粒沉积于动脉壁内膜,胆固醇晶体可上调斑块中NLRP3炎症小体成分的表达[27]。正常生理状态下,巨噬细胞通过新生高密度脂蛋白(high density lipoprotein,HDL)促进胆固醇逆向转运并进入肠肝循环,HDL可溶解部分胆固醇晶体并抑制巨噬细胞中胆固醇诱导的NLRP3炎症小体活化及促炎因子IL-1β分泌[32]。持续性炎症反应会导致HDL功能障碍,尤其是胆固醇外排能力降低,这将进一步加剧胆固醇结晶形成和细胞死亡。超负荷的胆固醇晶体在巨噬细胞摄取及降解过程中会造成溶酶体损伤及自噬功能障碍,促进NLRP3炎症小体激活及促炎因子IL-1β的产生和分泌[33]。胆固醇晶体通过刺激中性粒细胞外陷阱(NETs)形成,进一步触发病变斑块区域内巨噬细胞中NLRP3炎症小体激活[34]。此外,胆固醇晶体还通过调控氧化应激转录因子Nrf2相关机制介导NLRP3炎症小体的激活[35]。胆固醇结晶除具有机械和毒性作用,还通过引发炎症反应来诱导动脉壁损伤,诱导细胞死亡并造成纤维帽穿孔,出现斑块破裂、出血及血栓形成等一系列变化[28]。总体来讲,脂质与NLRP3相互作用诱导不可逆转的炎症反应是动脉粥样硬化病变的基础。
5 TET2突变与NLRP3炎症小体
老化相关的体细胞突变体在高龄人群中普遍存在,并且体细胞突变介导的克隆性造血参与动脉粥样硬化性疾病的发生和发展[36]。正常衰老与造血系统中体细胞突变积累相关,体细胞突变赋予其一定的竞争生长优势进行异常自我更新和髓系扩张,即克隆性造血[37]。不确定潜质的克隆性造血(CHIP)在老年人群中最为常见(65岁以上人群中>10%),又被称为与年龄相关的克隆造血(ARCH),涉及已识别的癌症驱动基因TET2和DNMT3A的突变[38]。TET2突变诱导的CHIP是新的心血管疾病危险因素[39],这与部分携带TET2突变的炎症性巨噬细胞有关,TET2在动脉粥样硬化进程中发挥重要作用[40]。
在TET2-/-小鼠的骨髓巨噬细胞中,oxLDL和内毒素导致溶酶体功能障碍并促使CXC家族趋化因子分泌增加,增强单核细胞和其他血细胞向炎症反应区域募集。oxLDL作用于内皮细胞抑制TET2蛋白表达,协同增加NF-κB/NLRP3活化,促进Caspase-1和IL-1β成熟及分泌,参与动脉粥样斑块的形成[41](图1)。研究显示,造血系统或髓样TET2缺乏导致心脏重塑和功能恶化,并伴有促炎因子IL-1β表达增加,选择性NLRP3炎症小体抑制剂治疗可预防心力衰竭的发展[42]。体细胞突变所致CHIP与冠心病或早发性心肌梗死的风险增加有明显相关性[43]。TET2通过招募HDAC2调控IL-6基因启动子从而抑制炎症持续发展,在炎症后期反馈抑制IL-6的分泌[44]。TET2通过抑制JAK/STAT信号通路的负调控因子mRNA合成,促进髓系细胞的生成及免疫细胞增殖,从而使机体针对病原微生物的入侵及时进行免疫反应[45]。CHIP恶化小鼠动脉粥样硬化,增加IL-6及IL-1β的表达[46]。Kaasinen等[47]则认为,在老年人体内出现TET2突变克隆反映出对衰老的抵抗力增强,TET2与NLRP3炎症小体的典型或非典型激活剂有关。TET2突变介导的CHIP通过NLRP3/IL-1β炎症通路途径,影响动脉粥样硬化进展[48]。为模拟TET2介导的克隆性造血作用,利用混合骨髓嵌合体策略来改造小鼠,斑块中招募少量的TET2突变细胞足以激活NF-κB/NLRP3炎症小体途径来驱动血管炎症,而NLRP3抑制剂(MCC950)可显著改善ApoE-/-小鼠体内由TET2体细胞突变介导的动脉粥样硬化[49]。总之,NLRP3炎症小体在TET2突变驱动的动脉粥样硬化中的重要作用,同时上调TET2表达亦是治疗动脉粥样硬化的新策略。
6 NLRP3炎症小体作为动脉粥样硬化治疗的新靶点
血浆胆固醇水平升高,尤其是低密度脂蛋白胆固醇(LDL-C)被认为是动脉粥样硬化的首要危险因素。因此,以降低LDL为目标的他汀类药物是目前治疗AS的主要举措。虽然他汀类药物可通过干预NLRP3蛋白的表达从而减少炎症因子的释放并减轻动脉粥样硬化病变,但是血液胆固醇水平下降的大部分人群中动脉粥样硬化病变仍有很大程度发展。因此,单纯依靠降脂治疗不能够降低所有患者未来发生心血管事件的风险。2-羟丙基-β-环糊精(CD)作为胆固醇晶体溶解剂,不仅可以减少动脉粥样硬化病变并减轻胆固醇负荷,而且能够诱导胆固醇外排并促进动脉斑块溶解及消退[50]。胆固醇晶体消融疗法适用于预防或临床治疗人类动脉粥样硬化,有望提供一种替代方法来稳定易损斑块。
CANTOS实验表明[51],康纳单抗(Canakinumab,靶向IL-1β的治疗性单克隆抗体)在不影响血脂水平的前提下,可减少促炎因子产生及释放并降低心血管疾病的风险。在动脉粥样硬化方面,NLRP3炎症小体显示出关键作用及治疗潜能。现阶段,针对NLRP3炎症小体正进行各种基础研究和临床试验,以评估抗炎药对动脉粥样硬化的治疗效果。数据显示[52],MCC950是一种有效的选择性NLRP3抑制剂,可抑制LPS和胆固醇刺激时巨噬细胞及树突细胞中IL-1β的成熟和分泌,改善动脉粥样硬化病变发展。降糖药物二甲双胍可通过抑制AMPK激活和调节TRX-1/TXNIP表达,减弱NLRP3炎症小体的激活效应,抑制糖尿病模型中动脉粥样硬化的进展[53]。此外,靶向TET2突变体和其他CHIP及其炎症环境的基本原理和前景,作为抑制CHIP相关动脉粥样硬化性疾病的潜在手段。携带TET2体细胞突变的个体可能比普通人群采用NLRP3炎症小体靶向治疗获益更大,这将为TET2体细胞突变的动脉粥样硬化患者的精准个性化治疗提供基础。围绕NLRP3炎症小体展开的动脉粥样硬化研究为冠心病及卒中等疾病治疗提供新策略,其临床使用效率仍需要进一步探索。
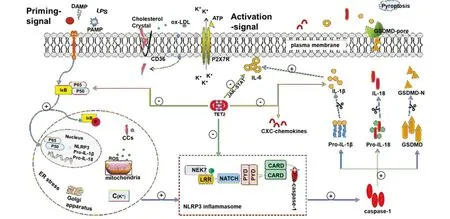
oxLDL、ROS等代谢因子激活NLRP3炎症小体,活化的Caspase-1切割下游因子IL-1β及GSDMD等前体分子,随后促炎因子经由GSDMD-pore释放并加速动脉粥样硬化进程。TET2抑制IκB磷酸化及NLRP3炎症小体活化,影响IL-1β等促炎因子和CXC趋化因子成熟及释放。此外,TET2经由JAK-STAT通路负反馈调控IL-6表达。图1 NLRP3炎症小体激活及抑制途径Fig.1 Activation and inhibition of the NLRP3 inflammasome
7 小结与展望
近年来,有关动脉粥样硬化研究进展较快,尤其是NLRP3炎症小体在动脉粥样硬化发生发展中的作用方面。NLRP3炎症小体参与动脉粥样硬化病变过程中的免疫应答及炎症反应,介导全身性炎症反应可能导致机体免疫稳态失衡,因此靶向NLRP3炎症小体减少、促炎因子的成熟及分泌并减弱其生物学效应,被视为AS预防及治疗的新方向。目前,关于脂质介导的NLRP3炎症小体活化在动脉粥样硬化中作用的研究已经有所进展,但有关于TET2介导的克隆性造血在炎症通路中作用的研究才刚开始。我们不仅需要探索NLRP3炎症小体具体的激活机制以及体细胞TET2突变对DNA甲基化及克隆性造血的作用,还需进一步研究NLRP3炎症小体与TET2及其他体细胞突变的交互作用,从而加深我们对动脉粥样硬化的发病机制的理解,为动脉粥样硬化性心血管疾病的临床诊断、预防及治疗提供新策略。
猜你喜欢
中西医结合心脑血管病杂志(2022年19期)2022-11-19
中国临床解剖学杂志(2022年1期)2022-11-15
材料与冶金学报(2022年2期)2022-08-10
检验医学与临床(2022年12期)2022-06-27
中国人兽共患病学报(2022年5期)2022-06-08
当代陕西(2022年5期)2022-04-19
科学与财富(2021年33期)2021-05-10
现代装饰(2021年1期)2021-03-29
新农业(2020年22期)2020-12-18
今日农业(2020年24期)2020-12-15
