
可见光促进二氧化碳参与的羧基化反应
2022-08-06 04:37张琴芳余达刚
高等学校化学学报 2022年7期
张 振,邓 煜,张琴芳,余达刚
(1.成都大学食品与生物工程学院,成都 610106;2.四川大学化学学院,绿色化学与技术教育部重点实验室,成都 610064)
近几十年来,温室气体二氧化碳(CO2)受到了广泛关注. 另一方面,CO2具有无毒、廉价易得、储量丰富且可再生等特点,是合成化学中的理想C1合成子. 利用CO2参与有机反应,可以合成多种高附加值产品,变废为宝,具有重要意义[1~13].
在CO2参与的众多有机转化中,CO2参与的羧基化反应一直被广泛研究[14~20]. 羧酸类化合物广泛存在于天然产物、药物、日化品及工业原料中,是人类日常生活中不可或缺的一大类化合物,如食醋中的乙酸、橄榄油中的油酸、奶油中的丁酸、肥皂、化妆品及染料等与羧酸类化合物有千丝万缕的关系,此外,解热镇痛药阿司匹林和止痛药布洛芬等都具有羧酸这一重要的官能团. 通过合理设计,CO2可以与众多廉价易得的工业原料反应,通过一步法得到结构多样的羧酸类化合物. 尽管该领域已经取得了很多进展,但依然存在一些问题和挑战:首先,CO2具有较高的热力学稳定性,反应活性低,通常需要高温高压等苛刻条件;其次,CO2作为气体,在反应体系中的浓度相对较低,而且是非极性分子,与过渡金属催化剂作用力较弱,表现为动力学惰性;第三,羧酸化合物容易发生的脱羧反应是羧基化反应的逆反应,而且是一个熵增的过程,有可能导致羧基化反应效率不高,容易发生质子化等副反应.
近年来,光化学快速发展,为上述问题的解决提供了很好的突破口[21~31]. 光能是一种取之不尽,用之不竭的绿色能源. 在资源、环境问题日益严重的当下,光能的高效利用备受关注. 光化学体系通过吸收光子能量,将光能转化为化学能,能够在温和条件下形成高活性中间体,在促进惰性键和小分子活化领域具有重要应用前景. 随着可见光化学的发展,可见光促进的CO2转化受到了关注. 近年来,研究人员在温和条件下利用可见光实现了CO2参与的一系列底物[如烯烃(C=C)、炔烃(C≡C)、醛酮(C=O)、亚胺(C=N)、含C—X(X=卤素、氮、氧)等化合物、烃类]的羧基化反应(Scheme 1)[32~39]. 其中,可见光促进CO2参与烯烃(C=C)、炔烃(C≡C)、醛酮(C=O)、亚胺(C=N)和含C—X(X=卤素、氮、氧)等化合物的还原羧基化反应被广泛研究,通常需要使用三级胺类、甲酸(盐)和汉斯酯作为电子给体. 此外,研究者也在氧化还原中性条件下实现了可见光促进CO2参与的烯烃、炔烃及C(sp3)—H键的羧基化反应.

Scheme 1 Visible light⁃driven carboxylation with CO2
从整体上看,为提高反应的实用性,此类反应多数被设计在可见光下进行(通常采用蓝光,波长处于400~450 nm),而体系中参与反应的大多数化合物在可见光下无明显吸收,通常需要引入在可见光波长范围内有吸收的光催化剂. 在均相体系下,这些光催化剂一般采用小分子有机物(包括金属有机络合物和有机小分子染料)(Scheme 1),光催化剂在吸收可见光后被激发为激发态,激发态光催化剂具有较高的活性,部分可以通过单电子转移与体系中特定的物质发生反应,产生活性物种,进而推动整体反应的进行. 其中,涉及到还原羧基化反应的例子中,反应还需要引入还原剂(电子供体). 还原剂一般为富电子化合物(如胺类化合物),容易还原猝灭激发态光催化剂,得到还原态的光催化剂进一步参与反应. 另外,为使反应顺利进行,反应有时需要引入过渡金属化合物活化底物(如一些常用的含镍、钯、铑的催化剂),通过这些催化剂与光催化剂协同催化,最终实现底物的羧基化反应. 当然,个别情况下,反应体系中无需外加光催化剂,可能是底物(如汉斯酯)本身可以吸光,也可能通过贫电子物种和富电子物种形成可以吸光的复合物. 值得强调的是,虽然一些非均相催化剂[如使用较多的CdS光量子点、金属簇状化合物、金属有机框架材料(MOF)、石墨烯及氮化石墨烯等]在光催化二氧化碳转化中表现优异,并且可以被反复利用且种类丰富,因此用来实现羧基化反应同样具有非常大的潜力[40~47],但受篇幅限制,不在本文详细阐述.
基于以上内容,本文按不同底物类型,分类介绍和总结近几年来可见光促进CO2参与的羧基化反应研究,其中介绍的反应主要集中于均相有机光反应体系,并重点介绍反应特点和机理,同时对该方向发展前景进行了展望.
1 可见光促进二氧化碳参与的C=C烯烃类、C≡C炔烃类化合物羧基化反应
在有机合成中,烯烃是重要的合成前体,通过对烯烃的官能团化可以构建多种重要结构[48,49]. 对于可见光促进CO2参与的烯烃羧基化反应,通过不同的策略可以实现氢羧基化反应、双羧基化反应及其它双官能团化反应.
1.1 C=C烯烃类化合物的氢羧基化反应
自1978年Lapidus等[50]开始探索铑和钯催化乙烯与CO2的氢羧基化以来,CO2参与过渡金属催化的不饱和键的氢羧基化反应得到了较大的发展[51~63]. 由于烯烃化合物的氢羧基化反应需要用到较多的电子给体,而这些电子给体多为一些金属单质或有机金属试剂,由此可能产生一系列的安全和环境问题. 2017 年,Iwasawa 等[64]首次报道了可见光/金属铑协同催化CO2参与的烯烃氢羧基化反应. 与单纯依靠过渡金属催化不同,该过程采用安全廉价的iPr2NEt作为电子最终给体,反应需要加入Cs2CO3来抑制质子化产物和烯烃聚合产物的生成. 为促进还原羧基化的过程,该体系需要加入含缺电子基膦配体络合物. 该类反应产物具有较好的区域选择性,可以得到单一的α-羧基化产物(Scheme 2).

Scheme 2 Visible light photoredox/Rh dual catalysis for hydrocarboxylation of alkenes with CO2
该反应过程可能经历如下步骤:(1)光催化活性中间体RhH物种形成;(2)随后发生烯烃迁移插入,生成苄基铑络合物;(3)CO2插入该络合物的C—Rh键之间形成中间体羧基铑物种,此过程伴随光敏剂Ru络合物的能量转移;(4)光催化下发生质子与电子的转移,形成Rh(Ⅲ)络合物;(5)还原消除得到目标羧酸,同时释放Rh(I)的氢化物,参与下一轮催化循环. 整个过程中,烯烃的电子效应及苄基铑络合物的相对稳定性使反应具有良好的化学选择性,可以生成单一的α-羧酸化合物.
为进一步缩短反应时间,提高反应收率,Iwasawa等[65]进一步对该反应体系进行改进,用BI(OH)H代替iPr2NEt作为电子最终给体,并取得了不错的效果.
在上述体系开发不久,König等[66]于2018年开发了可见光/镍络合物协同催化CO2参与的烯烃氢羧基化反应(Scheme 3). 与Iwasawa等[64]的工作不同的是,该转化除采用相对廉价的镍络合物和有机光敏剂外,还通过选用不同的金属配体,实现了不同区域选择性的羧酸的合成,以高的选择性分别得到α和β位置的两类羧酸. 从König等[66]给出的机理和解释来看,反应中还原态的光敏剂会通过SET过程将电子给到镍络合物从而促进反应发生,而自身光激发态通过还原猝灭过程可回到还原态以进入下轮催化过程. 当反应选择neocuproine作为配体时,反应生成的Ni(Ⅱ)金属氢化物种可以快速地与烯烃发生迁移插入反应,得到α-选择性的Ni(Ⅱ)金属络合物,进而经过一系列的转变得到α-羧酸;而选择1,4-bis(diphenylphosphino)butane(dppb)作为配体时,镍(0)络合物的电子云密度较高,可以很好地与二氧化碳和烯烃发生氧化环金属化,从而生成环镍(Ⅱ)金属物种,进一步被单电子还原和进攻CO2,则可以高选择性地得到β-羧酸.
以上转化均通过光/金属协同催化的模式,实现了CO2参与的烯烃氢羧基化反应. 2018年,Yu等[67]利用有机光敏剂4CzIPN与三级胺还原剂的简单体系,实现了烯酰胺类化合物的氢羧基化反应,并高效合成了多种具有挑战性和重要的非天然的α,α-双取代氨基酸及相关衍生物(Scheme 4). 值得指出的是,如果无光敏剂或者在黑暗条件下,烯酰胺类化合物则会在碱的促进下发生β-碳氢键羧基化反应[68]. 除烯酰胺外,该体系同样适用于亚胺类底物的氢羧基化反应. 该反应同样具有较好的区域选择性和化学选择性. 机理研究结果表明,可见光、光敏剂、碱和还原剂在反应中均起到了非常重要的作用. 结合其它亲电试剂参与反应的情况及氘代实验,作者认为通过可见光催化连续单电子转化(SSET)还原生成的α-季碳阴离子是重要的反应中间体,可以进攻二氧化碳或其它亲电试剂得到目标化合物.

Scheme 4 Visible light photoredox catalysis for hydrocarboxylation of enamides and imines with CO2
与以上CO2双电子活化过程不同,CO2单电子活化过程通过将CO2单电子还原得CO2自由基阴离子(CO·2−),可以进攻烯烃得到烯烃氢羧基化产物. 然而,由于CO2具有很高的化学稳定性,反应需要较高的能量,而且生成的CO2自由基阴离子活性较高,不容易控制其反应的选择性,因此该类转化在有机合物中较为少见,且更具有挑战性. 2017年,Jamison等[69]首次采用连续流动装置,在紫外光照射和光催化剂对三联苯催化下,得到二氧化碳自由基阴离子活性中间体,并实现了双键的β选择性加氢羧化反应. 直到2020年,Yu等[70]在其开发的硫羧基化反应的基础上,报道了第一例可见光驱动、无外光催化剂加入的丙烯酸酯、苯乙烯与CO2的氢羧化反应(Scheme 5)[71],从而实现了不寻常的选择性(得到β-羧酸化合物). 值得指出的是,这种方法还适用于具有挑战性的三取代和四取代丙烯酸酯以及三取代苯乙烯. 此外,作者通过使用4-苯乙基取代的Hantzsch酯(4-PE-HE)作为有效还原剂,首次实现了可见光驱动硫醇催化的丙烯酸酯和苯乙烯的氢羧基化反应. 通过机理实验推测,硫醇盐和丙烯酸酯/苯乙烯之间可以形成电子转移复合物(CTC),该络合物在可见光激发下还原CO2得到CO2自由基负离子,CO2自由基负离子与烯烃反应得到目标羧酸,而硫自由基被还原剂还原进入催化循环. 相关实验显示,这种还原反应条件下也可能会产生烯烃和CO2自由基阴离子. 该体系局限于活化烯烃,不能实现非活化烯烃的选择性加氢羧化. 进一步的密度泛函理论(DFT)计算表明,硫醇盐与脂肪族烯烃之间形成相应的CTC的可能性较低.
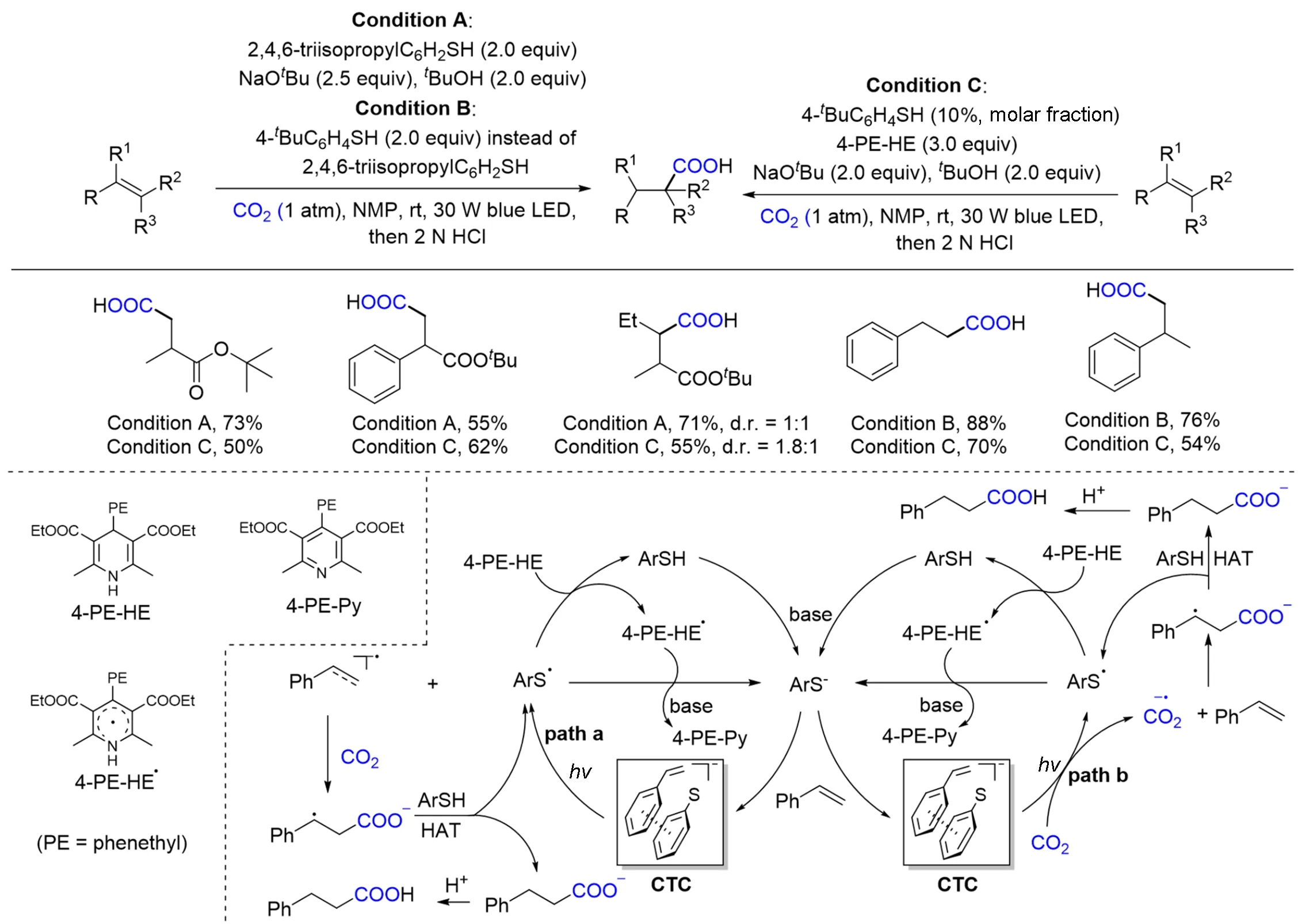
Scheme 5 Visible light⁃driven photocatalyst⁃free hydrocarboxylation of acrylates and styrenes with CO2
以上是目前主要报道的可见光促进二氧化碳参与的烯烃类化合物氢羧基化反应研究. 虽然研究人员通过不同的策略实现了该类转化反应,且收率和选择性都取得了较好的结果,但目前此类转化主要集中于活化烯烃,需要进一步研究非活化烯烃的相应转化.
1.2 C=C烯烃类化合物的双羧基化反应
二酸化合物是一类重要的羧酸化合物,其结构的特殊性使其在高分子材料的合成与开发中有着重要的应用. 近年来,科研人员围绕二酸的合成开发了很多有价值的反应体系[72~74]. 其中,以CO2为羧基源合成二酸类化合物主要集中在电化学领域[75~77],光催化下制备二酸化合物的报道较少且有一定的挑战性. 2021年,Yu等[78]首次报道了光催化下,二氧化碳参与烯烃的双羧基化反应(Scheme 6). 机理研究表明,烯基自由基负离子是反应的关键中间体,反应经由烯烃的两次单电子还原(SSET),两次进攻CO2来生成丁二酸类化合物. 该反应的底物适用性广,烯烃、二烯、联烯、稠环芳烃和杂芳烃均可以通过此方法得到相应的二烯化合物. 此外,官能团兼容性高、能放大至克级规模和产物易于衍生化等也是该反应的优点.
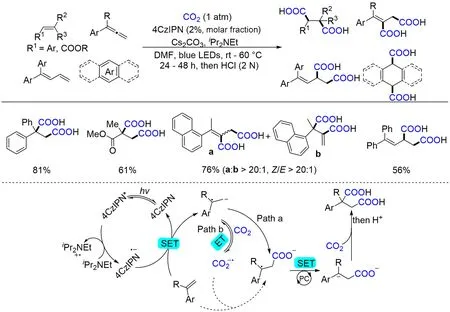
Scheme 6 Visible light⁃driven dicarboxylation of alkenes,allenes,and(hetero)arenes with CO2
1.3 C≡C炔烃类化合物的氢羧基化反应
炔烃也是重要的工业原料之一. 通过炔烃的氢羧基化反应,可以得到重要的α,β-不饱和羧酸.1982年,Hoberg等[79]首次实现了炔烃和CO2在Ni(0)和还原剂存在下反应生成五元金属环,进一步经酸处理后制备了丙烯酸混合物. 随后,科学家们开发了多种金属催化/介导的利用CO2作为C1源的炔烃加氢羧化反应[80~82]. 然而,通过可见光促进的炔烃氢羧基化反应具有一定的挑战性,其原因在于光催化下多为自由基反应,由此带来了较难的化学、区域、Z/E选择性控制. 2018年,Wu等[83]采用光/钴协同催化的策略,实现了多种非端炔的氢羧基化反应,并以较好的选择性和收率得到一系列α,β-不饱和羧酸,而对于脂肪端炔,则可以发生关环反应生成多种杂环化合物(Scheme 7). 机理研究表明,光催化剂可以促进单电子过程和烯炔异构化过程. 作者认为该反应是由炔烃、CO2和Co(Ⅰ)物种的氧化环金属化形成钴杂五环内酯的关键中间体引发的. 当使用末端炔烃时,另一个炔烃迁移插入到钴杂五环内酯中,得到2-吡喃酮. 非端炔烃则进行质子化得到氢羧化产物. 有趣的是,芳环上的辅助取代基经过一锅炔烃加氢羧化、烯烃异构化和环化形成有价值的杂环,包括2-吡喃酮、香豆素、2-喹诺酮、2-苯并氧杂环庚酮和γ-羟基丁烯内酯.
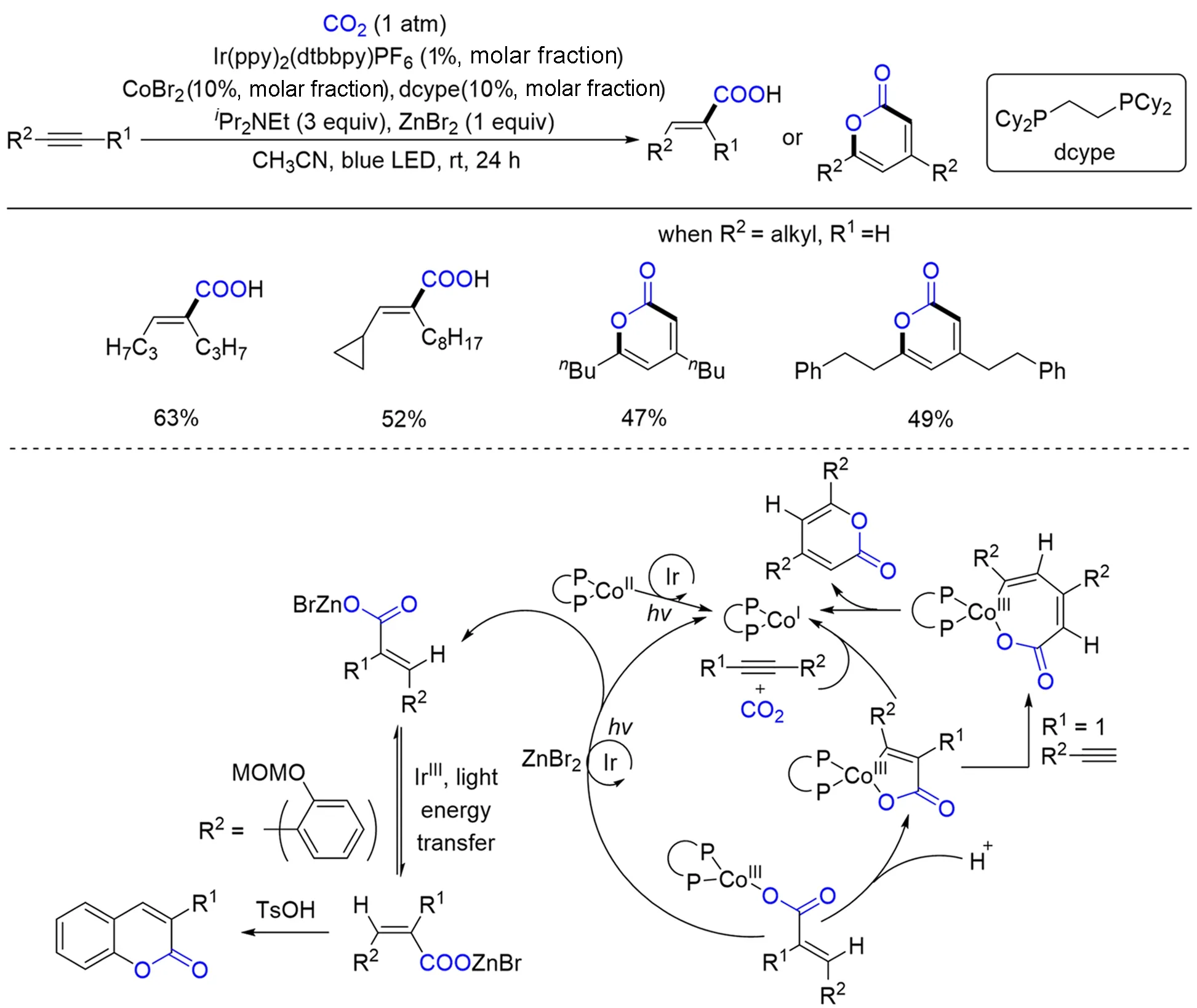
Scheme 7 Visible light photoredox/Co dual catalysis for hydro⁃/carbocarboxylation of alkyne with CO2
2021年,Fu等[84]通过密度泛函理论计算进一步完善了Wu等[83]推测的机理(Scheme 8):发现最可行的催化循环步骤为催化剂Ir的光激发和还原淬灭、H转移、羧化及催化剂再生;H转移是决定速率和区域选择性的步骤;非金属还原剂iPr2NEt在反应中的作用包括还原猝灭过程中的电子供体、电子受体和氢化物耦合电子转移(HCET)步骤中的氢化物供体;同时该工作也对反应的选择性作了解释.
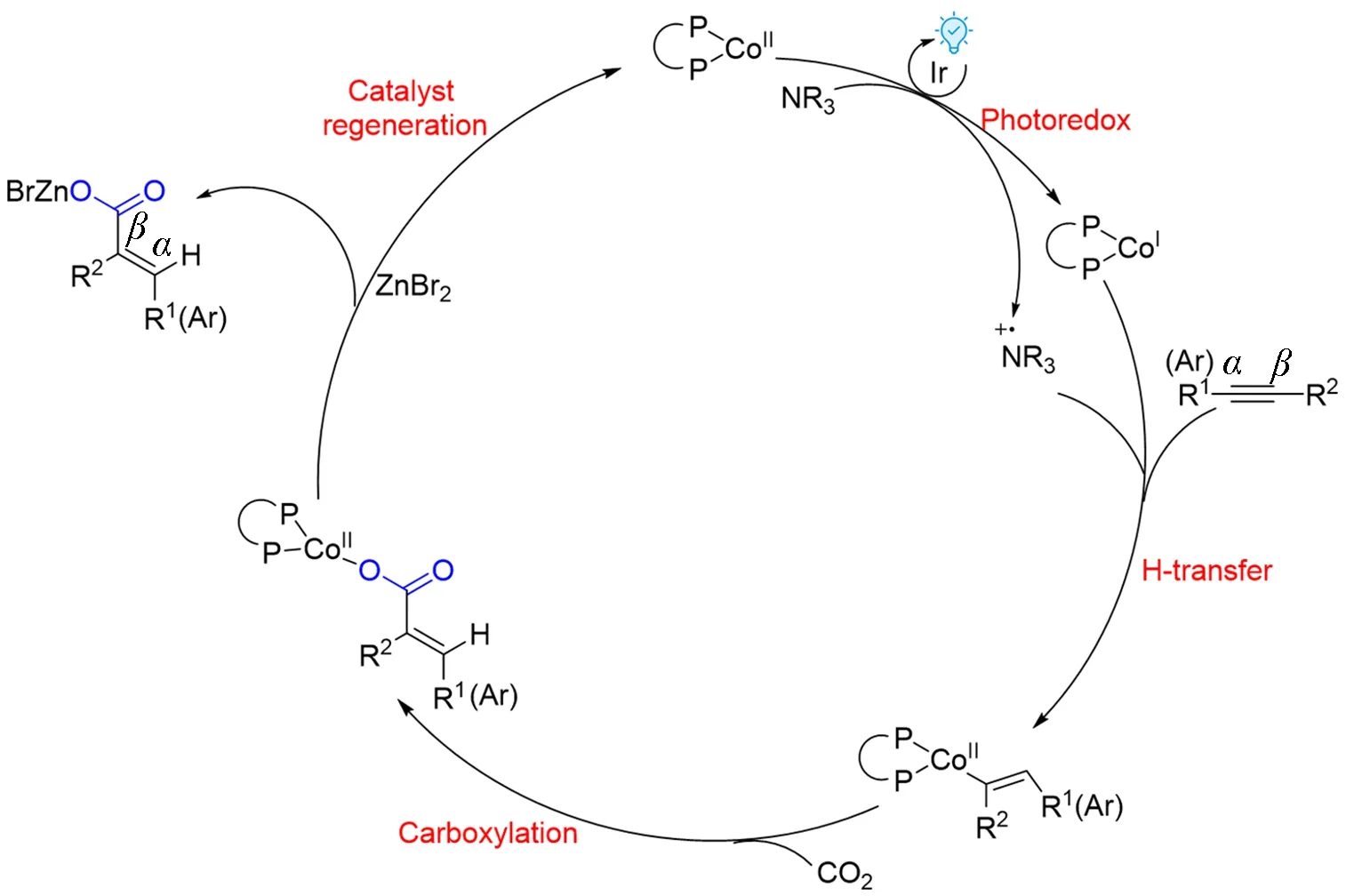
Scheme 8 Mechanistic study on visible light photoredox/Co dual catalysis for hydro⁃/carbocarboxylation of alkyne with CO2
2 可见光促进二氧化碳参与的含C—X(X为卤素,氧,氮等)化合物羧基化反应
2.1 可见光促进二氧化碳参与的含C—X(X为卤素)化合物的羧基化反应
有机(类)卤代烃类化合物的合成方法繁多,来源广泛,是一大类重要的化工原料和有机中间体.通过(类)卤代烃与CO2反应合成羧酸,无论从原料的易得性角度上讲,还是从产物的重要性角度上讲,都非常重要. 人们先后通过将(类)卤代烃制备成格氏试剂或是采用高活性高敏感的金属有机试剂与(类)卤代烃反应,得到可以与二氧化碳反应的有机活性中间体制备羧酸. 但受限于官能团兼容性差、实验操作不便等问题,科学家又陆续将目光转向过渡金属催化的方式,并取得了令人瞩目的成就[17~20]. 然而,由于这类反应往往需要加入金属单质或有机金属试剂,给反应的操作和后处理带来了不便,从而限制了此类反应的应用. 考虑到光催化还原过程,不仅可以利用更为绿色的有机电子给体作为还原剂,并且反应活性更高、条件更为温和,所以近年来研究人员逐步将(类)卤代烃类化合物的羧基化反应的探索重点由过渡金属催化转向光催化.
2017 年,Iwasawa 和Martin 等[85]首先在这方面取得了突破. 通过光/钯协同催化的策略,以三级胺作为最终电子给体,金属铱络合物为光催化剂,实现了二氧化碳参与的芳基卤代物的还原羧基化反应,并得到了一系列芳基羧酸(Scheme 9). 芳基溴代物和芳基氯代物均可以很好地参与反应,但需要不同的金属配体. 机理研究表明,碳卤键对Pd(0)L氧化加成形成关键中间体ArPd(Ⅱ)XL,进而与CO2反应形成络合物ArCOOPd(Ⅱ)XL,随后被还原态光敏剂单电子还原生成ArCOOPd(Ⅰ)XL,并进一步被单电子还原得到目标羧酸,同时释放LPd(0)进入下一轮循环. 另外,ArPd(Ⅱ)XL也可能首先与CO2络合形成ArPd(Ⅱ)XL(CO2),进而再通过单电子还原得到ArPd(Ⅰ)XL(CO2),此时CO2插入C—Pd 形成ArCOOPd(Ⅰ)XL并进一步被还原得到最终产物.

Scheme 9 Visible light photoredox/Pd dual catalysis for carboxylation of aryl halides with CO2
几乎同时,König[86]也实现了光/镍络合物协同催化卤代烃化合物的还原羧基化反应(Scheme 10).该反应中以4CzIPN 为光敏剂,HE 为最终电子给体,同时加入neocuproine 作为金属配体,类似该小组之前报道的氢羧基化反应体系[66]. 反应中加入的碳酸钾不仅起到碱的作用,也是羧基来源之一,CO2氛围则能够提升反应效率. 从作者给出的机理循环来看,整个过程先发生氧化加成过程形成中间体Ni(Ⅱ)络合物,然后被单还原为Ni(Ⅰ)络合物,进而与CO2反应得到羧酸镍(Ⅰ)中间体,进一步发生单电子还原,得到目标羧酸的同时释放Ni(0)物种,参与下一轮催化循环. 光催化过程则通过单电子转移方式促进该还原羧基化反应进行. 在开发方法学的同时,作者也采用该方法合成了具有生物活性的MCPB,证明了其实用性. 值的一提的是,该体系不仅适用于芳基溴代物,对于烷基溴代物和三氟甲磺酸酯同样可以实现高效转化,从而极大地拓展了该方法的适用范围.
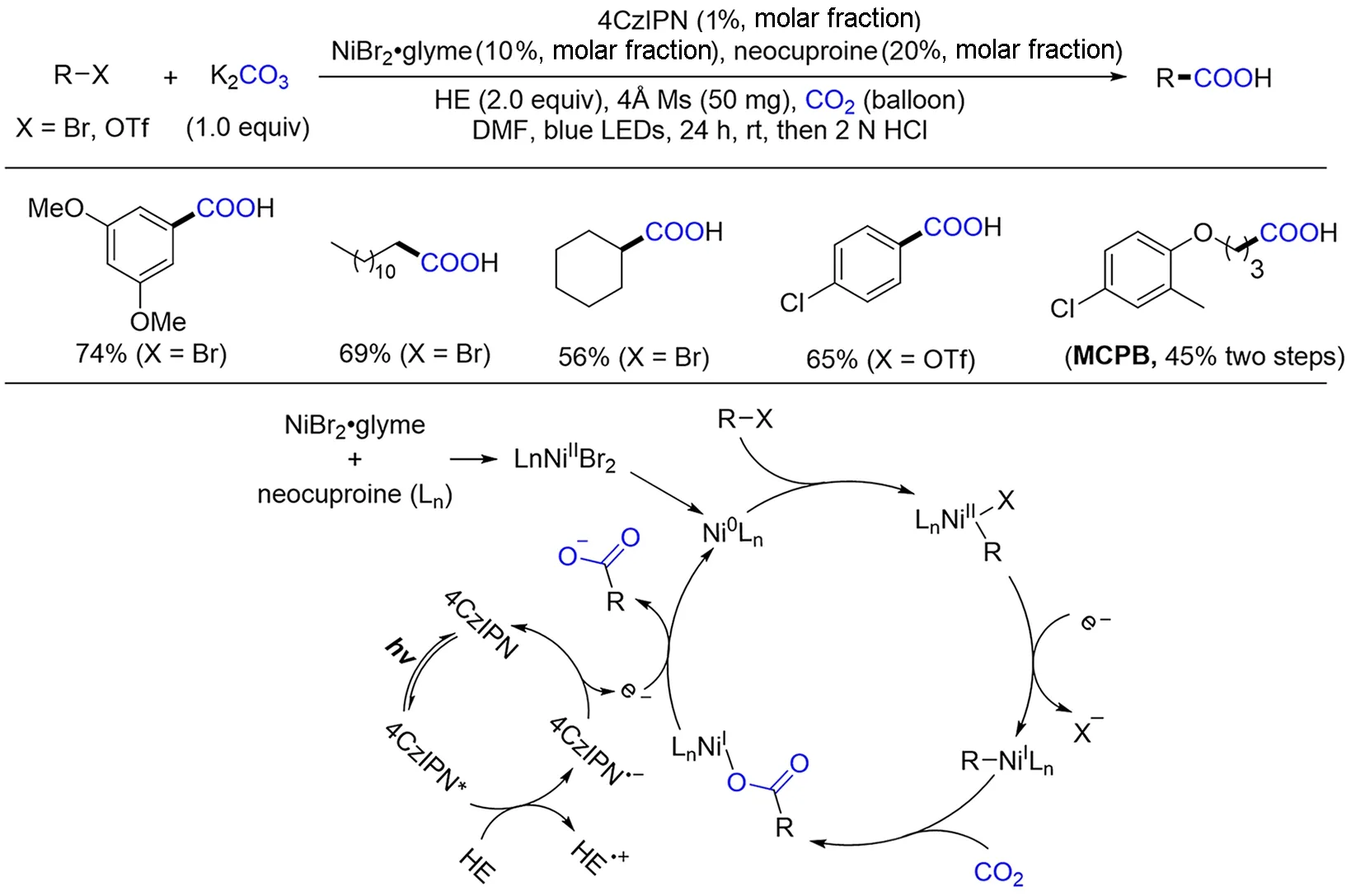
Scheme 10 Visible light photoredox/Ni dual catalysis for carboxylation of aryl bromides/triflates and alkyl bromides with CO2
同样是采用光/镍络合物协同催化反应,2019 年,Crespi,König 和Martin 等[87]则通过调控配体和碱,实现了一类独特的通过C(sp3)—X键远程活化C(sp3)—H键并实现CO2参与的羧基化反应(Scheme 11). 机理研究表明,C—X 键与金属氧化加成反应生成Ni(Ⅰ)络合物后,进一步发生β-H 消除生成烯烃,再次发生氢金属化反应,最终生成较为稳定的C—Ni键,进而与CO2反应实现羧基化反应.

Scheme 11 Visible light photoredox/Ni dual catalysis for remote carboxylation of sp3 C—H bonds with CO2
苄基卤代物是一类非常重要的化学中间体和化工原料,来源广泛且制备方法较多. 通过还原羧基化反应,苄基卤代物可以生成布洛芬等重要的羧酸类化合物——芳基乙酸及其相关衍生物. 之前科研人员已通过电催化、过渡金属催化或是制备格氏试剂的方式,实现了苄基卤代物的还原羧基化反应[88,89]. 2021年,Yu等[90]在无过渡金属的条件下,利用相对廉价易得的有机染料为光敏剂,有机胺为电子给体,实现了CO2参与的一系列苄位氯代物和溴代物还原羧基化反应(Scheme 12). 在该反应体系下,一级、二级和三级的苄位卤代物均可以较好地发生转化. 机理研究表明,底物经历了2次单电子还原(SSET)得到碳负中间体,进而亲核进攻CO2得到目标芳基乙酸化合物.
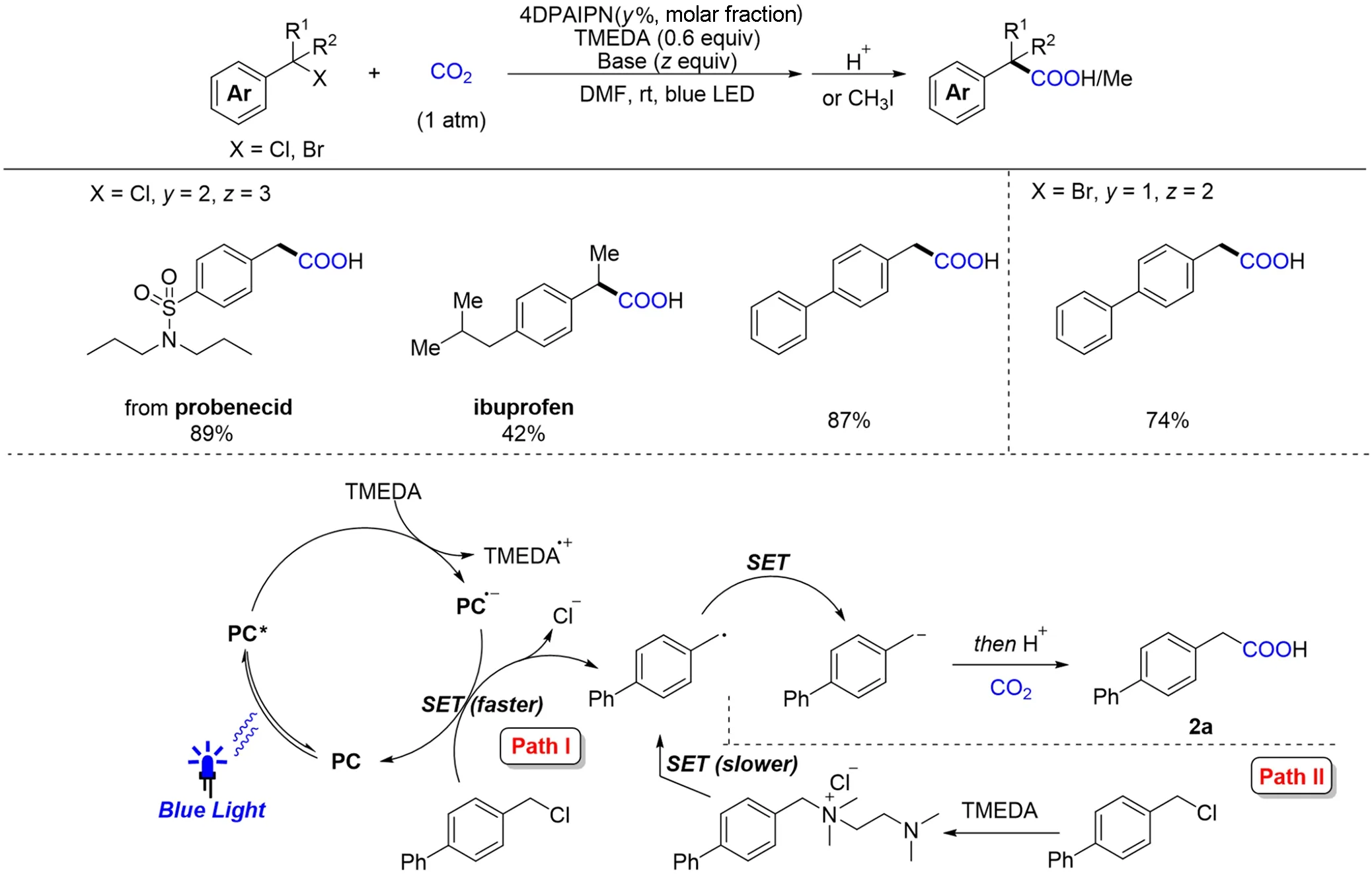
Scheme 12 Visible light photoredox⁃catalyzed reductive carboxylation of benzylic C—X(X=Cl,Br)bondswith CO2

Scheme 13 Visible light catalysis for carboxylation of C(sp2)—F bonds with CO2
与C—Cl和C—Br键相比,C—F键具有更高的能量而更难被活化,所以针对含C—F键化合物的羧基化反应更具有挑战性[91]. 2019年,Feng等[92]利用光/钯协同催化体系,首次实现了二氟烯烃C(sp2)—F键的还原羧基化反应,得到一系列的α-氟代羧酸[Scheme 13(A)]. 机理研究表明,C(sp2)—F键活化是通过还原态光敏剂的SET还原实现的. C(sp2)—F键断裂后,形成氟代烯基自由基并被Pd(0)物种捕获后形成Pd(Ⅰ)络合物中间体,进而与CO2反应形成羧酸Pd(Ⅰ)络合物,随后被还原态光敏剂单电子还原,得到目标氟代羧酸化合物的同时重生Pd(0)进入下一轮催化循环中. 虽然反应产物为Z/E的混合物,但选择性较好,作者推测其原因可能与Pd(Ⅰ)—烯基氟络合物存在一定程度的Z/E互变有关. 值的关注的是,同时期,过渡金属铜催化的C—F 键的羧基化反应得到了较好的发展,与光催化转化形成互补[93,94].
最近,Yu等[95]实现了更具挑战性的芳香C(sp2)—F的羧基化反应,实现了一系列多氟芳烃的选择性羧基化反应并得到相应的含氟羧酸类化合物. 该反应中,采用甲酸盐作为最终电子给体,甲酸盐通过HAT过程被氧化为二氧化碳自由基阴离子,该物种是还原多氟代芳烃的关键物种,可以将多氟代芳烃还原为氟代芳烃自由基,并进一步被还原态光敏剂还原,得到相应阴离子捕获二氧化碳,最终得到目标的含氟芳香羧酸类化合物. 而光敏剂被氧化后可进一步被可见光激发,氧化HAT试剂DABCO,再次攫取甲酸盐氢,推动反应进行[Scheme 13(B)].
除C(sp2)—F键的还原羧基化反应外,2021年,Yu等[96]实现了可见光催化多种C(sp3)—F键的还原羧基化反应(Scheme 14). 反应不仅实现了含单氟的C(sp3)—F的羧基化反应,同时也实现了选择性的二氟、三氟化合物的单羧基化反应,各种单、二和三氟烷基芳烃(包括未活化的芳烃)在温和且不含过渡金属的条件下顺利进行选择性羧化转化. 此外,α,α-二氟羰基化合物也能进行选择性C—F键官能化. 基于机理研究,作者认为CO2在该反应中起到了电子转移和电子受体的双重作用:CO2在该体系中先后经历氢负离子还原和HAT过程形成富电子的CO2自由基阴离子,由于该阴离子较强还原性,可对含氟底物单电子还原,最终完成C—F 键的断裂形成碳自由基,进而在光催化下形成碳负离子,进攻CO2得到选择性羧基化产物. 作者通过DFT计算验证了所提出的催化循环. CO2双功能性概念的提出也为后续相关的研究提供了参考.

Scheme 14 Visible light photoredox⁃catalyzed selective carboxylation of C(sp3)—F bonds with CO2
2.2 可见光促进二氧化碳参与的含C—O键化合物的羧基化反应
含C—O 键的化合物可以由醇类、酚类直接或间接衍生得到,原料可直接从天然产物或工业原料简单加工后得到,种类多样,廉价易得,因此含氧化合物通过还原羧基化反应将其转化为羧酸具有重要研究价值. 2019 年,Iwasawa[97]和Jana 等[98]几乎同时实现了三氟甲磺酸芳基酯的还原羧基化反应(Scheme 15). 反应机理类似于之前Iwasawa 小组[85]开发的芳基C—X 键的还原羧基化反应. 值得一提的是,Iwasawa等[85]主要通过改变配体的结构实现了三氟甲磺酸烯基酯的还原羧基化反应,并得到了一系列的α,β⁃不饱和羧酸类化合物,极大地拓展了反应的适用性.
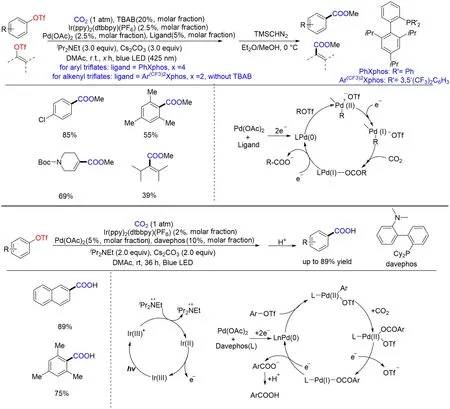
Scheme 15 Visible light photoredox/Pd dual catalysis for carboxylation of aryl and alkenyl triflates with CO2
在实现光催化C(sp2)—O还原羧基化的同时,研究人员也对同样重要且更具有挑战性的C(sp3)—O键的还原羧基化反应展开探索. Iwasawa等[99]在实现C(sp2)—O还原羧基化反应之后,进一步通过提升反应温度和改变过渡金属配体,实现了光/钯协同催化的各类苄醇酯(包括碳酸酯、羧酸酯、酰胺酯及膦酸酯等)的还原羧基化反应(Scheme 16). 控制实验结果表明,底物对金属钯的氧化加成为反应的决速步骤,而温度的提升则有利于这个过程. 光催化还原体系则与之前作用类似,用于反应中金属中心的还原. 反应可以通过配体和温度的调控,实现对芳基卤代物和苄基酯的选择性羧基化. 该反应机理类似于此前开发的C(sp2)—O键羧基化机理,此处不再赘述.

Scheme 16 Visible light⁃driven Pd⁃catalyzed carboxylation of activated C(sp3)—O bonds with CO2
几乎同时,Yu等[100]利用相对廉价易得的有机染料为光敏剂和有机胺为电子给体,实现了可见光催化一系列一级、二级及三级苄基醇(酯)的高效还原羧基化(Scheme 17). 其中,对于一级和二级苄基醇,可以通过一锅法形成酯中间体,一步将苄醇转化为羧酸,操作简单;而对于三级苄醇,一锅两步的方法可以有效提高反应效率. 此外,该方法也同样适用于各类α-羟基羧酸衍生物. 对比Iwasawa 课题组[99]采用的体系,该反应条件更加温合,且无需过渡金属参与,其原因在于反应中直接利用还原性较强的还原态光敏剂对苄位C—O键进行单电子还原,在室温下即可完成.
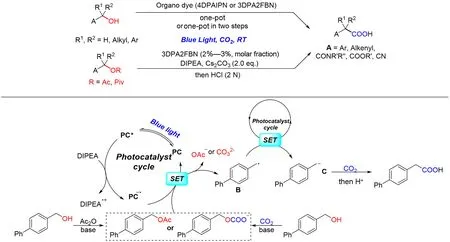
Scheme 17 Visible light photoredox⁃catalyzed transition metal⁃freecarboxylation of activated C(sp3)—O bonds with CO2
以上反应通常需要形成酯类化合物以提高底物中C—O 键的活性,从而实现羧基化反应. 最近,Xia等[101]采用可见光光氧化还原四苯基硼酸钠产生中性二苯基硼自由基活化游离醇的新策略,直接裂解游离醇的C(sp3)—OH键并与CO2发生交叉亲电偶联反应(Scheme 18). 该策略可以实现一系列伯、仲和叔苯甲醇的直接羧基化,为C—O键羧基化反应提供了更为高效绿色的思路.
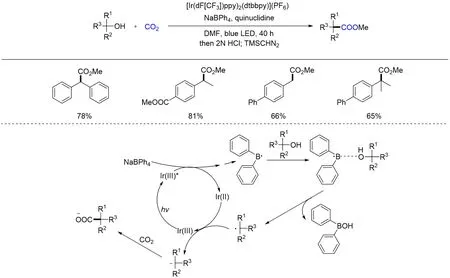
Scheme 18 Visible light photoredox⁃catalyzed carboxylation of free benzylic alcohols and CO2
2.3 可见光促进二氧化碳参与的含C—N键化合物的羧基化反应
除C—O键外,有机季铵盐中的C—N键还原羧基化反应同样是此领域研究的热点之一. 季铵盐结构稳定,易于得到和保存. 此前,通过过渡金属对C—N 进行活化,实现了相应偶联反应和羧基化反应[102~105]. 但值得注意的是,此类反应会产生副产物胺类化合物,但一般都被废弃,没有很好的利用.众所周知,有机胺是可见光催化中的一类重要电子给体. 基于此,2018年Yu等[106]结合光催化还原羧基化的特点,首次实现了无外加还原剂的苄基季铵盐的还原羧基化反应,一级、二级和三级苄基碳的季铵盐均可以很好地反应(Scheme 19). 机理研究表明,C—N键断裂后原位产生的胺作为电子给体参与催化循环反应. 之后,该类策略被成功应用于醛、酮及芳基腈的还原偶联中[107].

Scheme 19 Visible light photoredox⁃catalyzed carboxylation of tetraalkyl ammonium salts with CO2
3 可见光促进二氧化碳参与的C=O(醛酮)、C=N(亚胺)的还原羧基化反应
亚胺、醛和酮是合成复杂分子的重要中间体. 传统的合成方案通常利用羰基(C=O)和亚胺基(C=N)的碳-杂原子双键的强静电极化作用,使得碳原子带部分正电荷并以亲核加成反应为主导[108].极性反转反应可以改变这种天然极性,并在有机合成中开发非常规的成键策略,实现与非亲核试剂的反应[109,110]. 另一方面,氨基酸是一类重要且广泛存在的羧酸. 其中,α,α二取代-α-氨基酸在调节肽和蛋白质的结构、稳定性和功能方面发挥着重要作用. 2018年,Yu等[67]首次报道了可见光催化芳香亚胺经历偶极反转并与CO2的选择性加氢羧化,制备有价值的α,α二取代-α-氨基酸的反应(Scheme 4). 此反应在上文部分有所介绍,在此不在赘述.
随后,Walsh 等[111]报道了一例可见光或阳光介导的二芳基酮亚胺光氧化还原加氢羧基化反应.Cy2NMe一方面作为牺牲电子供体对亚胺光化学还原,偶极反转生成碳负离子,另一方面与产物作用生成羧酸盐沉淀,既能保护产物免于发生脱羧基化副反应,又能通过过滤提纯产物. 与余达刚课题组[67]采用有机催化剂及N-羰基亚胺底物不同,作者使用金属催化剂及N-烷基或N-芳基亚胺底物,2种方法在其它机制相同的情况下制备了具有不同N-保护基的α,α-二取代-α-氨基酸. 结合实验和自由基负离子中间体计算研究,作者推测的反应机理途径如Scheme 20所示. Cy2NMe还原猝灭被光激发的Ir(Ⅲ)生成胺自由基阳离子和还原性的Ir(Ⅱ),随后底物亚胺与Ir(Ⅱ)之间发生单电子转移生成胺自由基负离子并再生Ir(Ⅲ),α-胺基碳自由基负离子与胺自由基阳离子发生氢原子转移(HAT),生成亲核碳负离子中间体,然后与CO2反应生成最终产物.
值的一提的是,在光催化领域的反应中,与亚胺结构相近的酮(醛)肟酯类化合物也一直颇受关注,一些研究团队对环酮肟酯的自由基型开环反应展开了研究. 该类化合物通过单电子还原实现N—O键断裂形成亚胺自由基,进一步发生开环反应后可以得到一系列含氰基的自由基前体[112]. 利用这一特性,2020年,Yu等[113]首先实现了可见光促进的环酮肟酯的开环羧基化反应,并以中等到较好的收率得到一系列含氰基的羧酸类化合物. 由于氰基容易进一步转化,所以该反应可能在后续合成结构复杂的羧酸类化合物中得到应用. 机理研究表明,环酮肟酯自由基开环形成含氰基的自由基前体后,再一次经过单电子还原得到含氰基的碳负离子,并亲核进攻CO2,得到氰基取代的羧酸类化合物(Scheme 21).
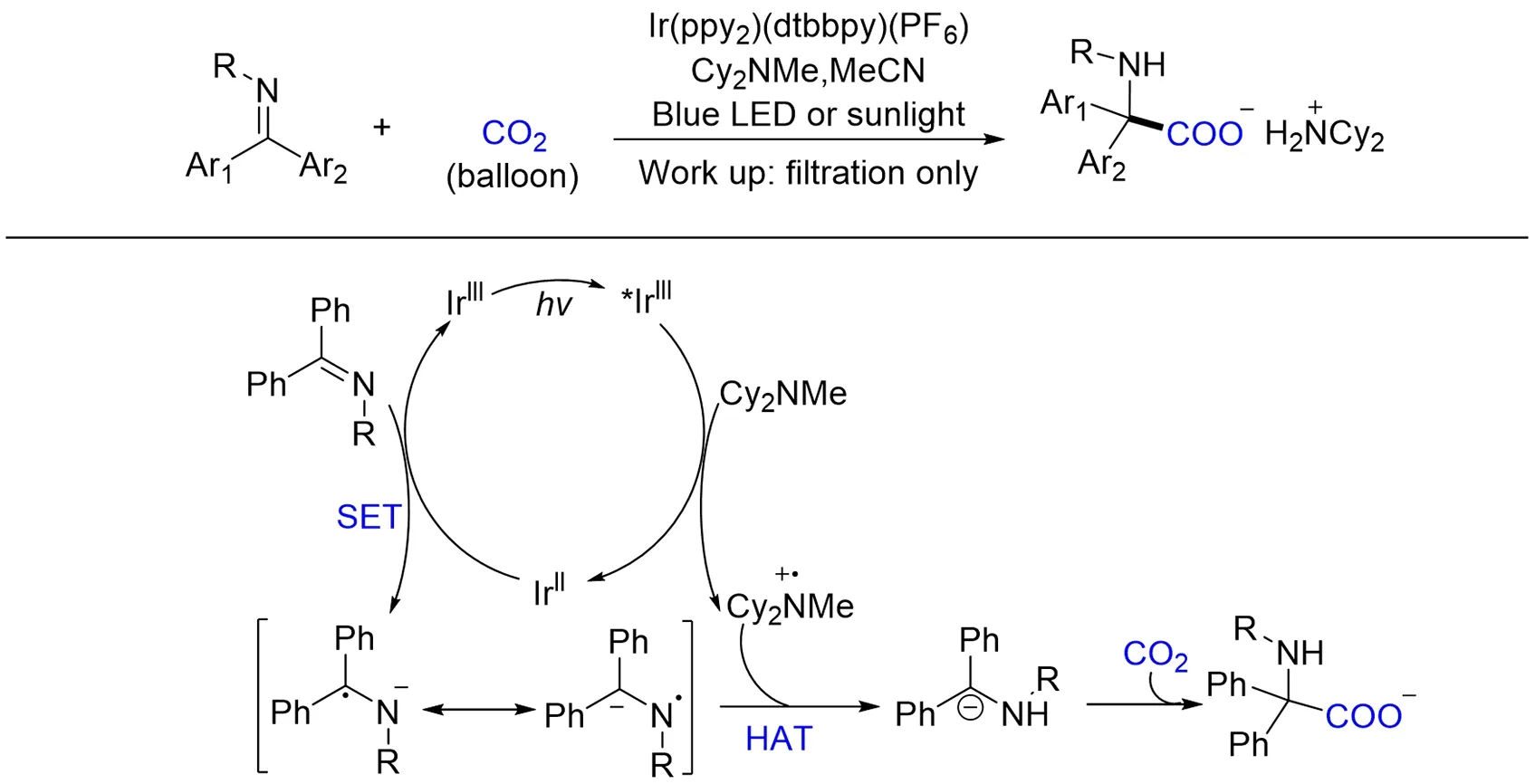
Scheme 20 Visible light photoredox⁃catalyzed hydrocarboxylation of imines with CO2
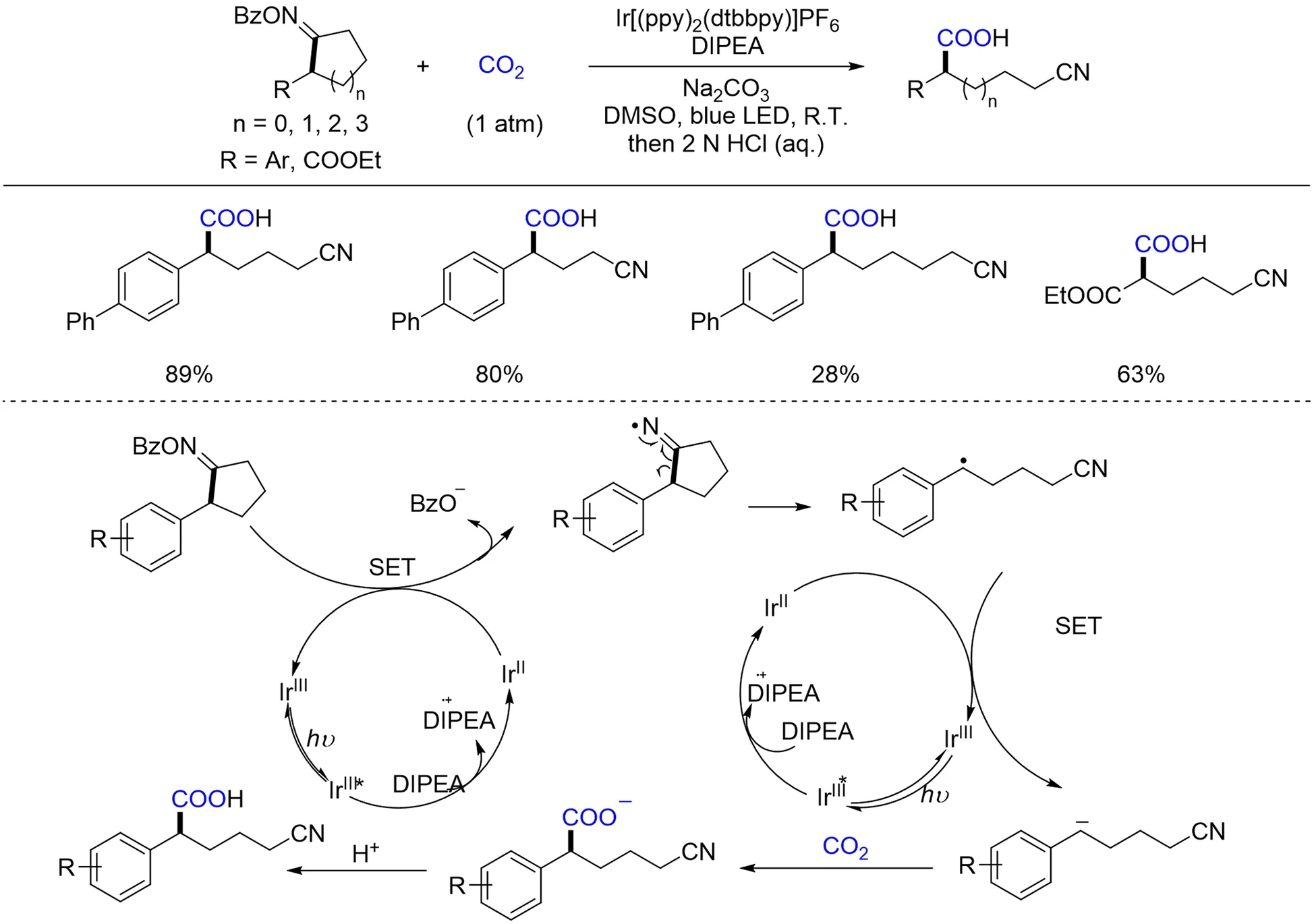
Scheme 21 Visible light photoredox⁃catalyzed ring⁃opening carboxylation of cyclic oxime esters with CO2
α-羟基羧酸是制药和聚合物工业中非常重要的结构. 然而,通过氰化和水解的传统合成方法需要使用有毒氰化物且条件恶劣. 近几十年来,以CO2为原料制备α-羟基羧酸的研究取得了重大进展[114,115],但大多数反应都存在使用化学计量的金属还原剂、官能团耐受性差、底物范围有限或发生竞争性副反应,尤其是频哪醇偶联和还原等缺点,从而阻碍了工业应用. Yu等[116]于2021年报道了一种使用路易斯酸性氯硅烷(TMSCl)作为活化和保护基团,利用可见光催化实现了不同羰基化合物偶极反转并与CO2发生羧基化反应(Scheme 22). TMSCl一方面作为路易斯酸活化羰基,促进可见光驱动的羰基的单电子还原,将羰基碳转化成易于与CO2反应的碳负离子;另一方面活化的氯硅烷可能充当临时保护基团以产生α-甲硅烷氧基碳自由基,增加自由基中间体的空间位阻,从而阻止频哪醇偶联副反应. 该策略对于各种羰基化合物(包括烷基芳基酮、二芳基酮、α-酮酰胺、α-酮酯和芳基醛)的羧化反应具有通用性和实用性,易于制备多种药物和天然产物的关键中间体. 该转化具有催化剂负载量低、选择性高、底物范围广、官能团耐受性好、反应条件温和(室温,1个大气压)以及易于克级反应等特点,为α-羟基羧酸的制备提供了很好的策略. 通过一系列的对比实验,作者推测了反应机理:被TMSCl活化的酮经历单电子还原产生相应的α-甲硅烷氧基苄基自由基,该自由基再经历一次单电子还原产生碳负离子,然后亲核进攻CO2生成相应的产物.
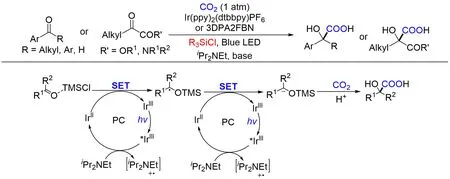
Scheme 22 Visible light photoredox⁃catalyzed umpolung carboxylation of carbonyl compounds with CO2
随后,Uozumi等[117]通过缓慢滴加反应物的方式,实现了羰基偶极反转羧化反应. 在光催化剂和还原剂DMBI存在下,不需要任何其它添加剂,芳香醛或酮与CO2反应形成α-羟基羧酸. DMBI作为双电子单质子供体,使得经历第一次单电子还原生成的酮基自由基氧负离子能够快速经历第二次单电子还原形成碳负离子,从而有效抑制自由基-自由基偶联副反应. 作者推测的反应机理如下:DMBI被可见光激发的Ir(Ⅲ)还原得到Ir(Ⅱ)和DMBI自由基阳离子,去质子化生成DMBI自由基. Ir(Ⅱ)还原芳族羰基化合物再生Ir(Ⅲ)光催化剂的同时产生酮基自由基氧负离子,进而在CO2促进作用下被DMBI自由基单电子还原生成苄基负离子,避免了自由基-自由基偶联副反应,并进而对CO2亲核进攻,然后进行质子化和脱羧,得到最终产物α-羟基羧酸(Scheme 23).
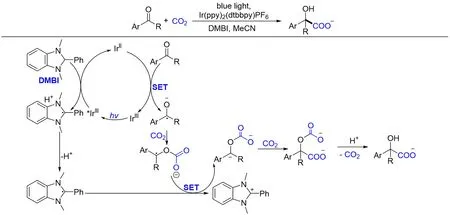
Scheme 23 Visible light photoredox⁃catalyzed carboxylation of aromatic aldehydes and ketones with CO2
4 可见光促进二氧化碳参与的去芳构化羧基化反应
芳香族分子属于最基本和最丰富的有机化合物类别之一,广泛应用于分子科学的所有领域. 去芳构化策略打乱了芳香族π系统平面结构,可快速获得不饱和的且通常是官能化的三维环状结构产品,这种直接的、从简单到复杂的合成逻辑在天然产物合成和小分子药物化学领域具有重要研究意义. 由光化学条件驱动的脱芳构化反应,尤其是利用丰富的可见光,被认为是一种以可持续和绿色的方式实现以前无法实现的转变的互补方式[118]. 1975年,Tazuke等[119]首次报道了光驱动菲去芳构化并以CO2作为羰基化试剂的羧基化反应,随后芳香族化合物与CO2的脱芳构化羧化反应取得了一定的研究进展[78,120,121].
在持续对二氧化碳参与的去芳构化领域关注的同时[122,123],2020年,Yu等[120]报道了一种可见光催化串联还原环化/交叉偶联方法,高选择性实现了吲哚与芳基卤化物和CO2两种亲电试剂的去芳基羧基化反应,有效避免了同位偶联、质子化、β-氢化物消除或异构化等副反应(Scheme 24). 该策略为合成难以获得的高价值的二氢吲哚-3-羧酸提供了一种有价值的参考. 机理实验推测,可见光激发的光催化剂被还原淬灭生成自由基阴离子,经历芳基溴的单电子转移回到基态,同时生成芳基溴自由基阴离子,断裂碳溴键生成的芳基自由基,并快速在吲哚的C2=C3双键上进行分子内自由基加成得到苄基自由基,实现吲哚杂环去芳构化,再经历一次光催化单电子还原生成的碳负离子,然后亲核进攻CO2实现羧基化反应.

Scheme 24 Visible light photoredox⁃catalyzed dearomative arylcarboxylation of indoles with CO2
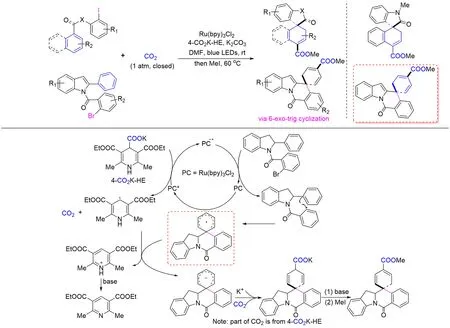
Scheme 25 Visible light photoredox⁃catalyzedreductive dearomative and spirocyclizative arylcarboxylation of nonactivated arenes with CO2
2021年,Li等[121]在温和的反应条件下通过自由基-极性交叉串联(RPCC)策略,对带有萘基、苯基和喹啉基的非活化芳烃进行可见光诱导的还原去芳构化,从而实现了芳烃与CO2的螺环化远程芳基羧化反应(Scheme 25). 这种还原去芳构化/芳基羧化方案可有效地从易得的芳香族前体制备有价值的三维羧酸衍生物,为构建复杂分子提供了一种独特的方法. 以2-苯基吲哚为底物,去芳构化主要通过6-exo-trig环化发生在未活化的苯环上,区别于Yu等[120]报道的通过5-exo-trig环化发生在活化的吲哚环的C2-C3双键上. 实验推测这种化学选择性去芳构化的机理途径如Scheme 25所示:可见光激发的光催化剂被4-CO2K-HE 猝灭得到自由基负离子和相应的二氢吡啶自由基,同时释放CO2,自由基负离子还原芳基溴化物产生芳基自由基并释放溴负离子,前者通过6-exo-trig 环化进行脱芳构化得到螺环自由基,进一步接受二氢吡啶自由基的单电子转移产生螺环阴离子中间体,其与CO2(部分CO2来源于4-CO2K-HE)的亲核加成产生羧酸盐,然后进行碱促进双键重排,最后经甲基化得到甲酯.
最近,Xi等[124]也报道了类似的可见光催化还原羧基化反应,发展了苄氧基邻卤代芳基醚的去芳构化反应,得到了一系列的螺环羧酸(Scheme 26). 反应具有非常高的化学选择性,可以高选择性得到含有共轭环烯的螺环羧酸,反应关键步骤为分子内自由基对苯环的加成及后续的还原过程.

Scheme 26 Visible light photoredox⁃catalyzedsequential dearomatization/carboxylation of benzyl o⁃halogenated aryl ethers with CO2
除了上述可见光诱导(杂)芳香烃分子内加成环化脱芳构化,实现与CO2的羧化反应外,Yu等[78]报道了一例特别的可见光驱动的(杂)芳香烃脱芳构化二羧基化反应,芳环脱芳构化并结合两分子CO2生成重要的二酸(Scheme 27). 该策略底物范围广,包括芳环上含有不同取代基的多种蒽衍生物、并四苯、苊、萘并[2,3-b]噻吩及吲哚衍生物等,反应条件温和,化学和非对映选择性较高,在有机合成、药物化学和材料科学中具有应用潜力. 该反应机理与前述的烯烃双羧基化(Scheme 6)[78]类似,这里不再赘述.

Scheme 27 Visible light photoredox⁃catalyzedreductive dearomative and dicarboxylation of polycyclic aromatic with CO2
5 可见光促进二氧化碳参与的C—H键的羧基化反应
C(sp3)—H活化一直是有机化学领域研究的一个重点和难点. CO2参与的C—H的羧基化反应不仅要解决C—H 键的活化问题,还要将其与CO2利用相结合,非常且有挑战性[125~127]. 2015 年,Masuda等[128]报道了紫外光促进o-烷基苯基酮与CO2的羧基化反应,并在2019年报道了利用紫外光促进CO2参与苯基和脂肪族C—H键的羧基化反应[129]. 这些工作都为CO2参与的C—H键直接羧基化反应提供了参考,是重要的发现,但是由于是紫外光促进的,这里就不多作介绍.
2019 年,König 等[130]报道了利用可见光促进氢原子转移(HAT)的光氧化还原策略,实现了CO2参与的苄基C—H键羧化反应(Scheme 28). 该反应在不添加任何金属试剂、牺牲电子供体、电子受体或化学计量添加剂的条件下得到相应的羧酸. 机理研究表明,该反应中的4CzIPN的氰基(CN)在光和硅基硫醇的作用下,被苄基取代生成4CzPEBN为主要的活性催化剂. 作者提出了苄基C—H键羧基化的催化循环,具体经历如下:(1)首先可见光激发下形成4CzPEBN*;(2)通过单电子转移,三异丙基硅硫醇(RSH)还原猝灭4CzPEBN*,产生硫醇自由基阳离子RSH•+,以及光敏剂的还原态,即4CzPEBN•-;(3)R—SH•+去质子生成亲电的硫基自由基R—S•;(4)R—S•从乙苯的苄基位置攫取一个氢原子后进入下一轮有机催化循环并产生苄基自由基;(5)之前形成的4CzPEBN•-还原苄基自由基形成苄基负离子.该负离子很容易被CO2捕获,最终生成羧酸化合物(Scheme 28).
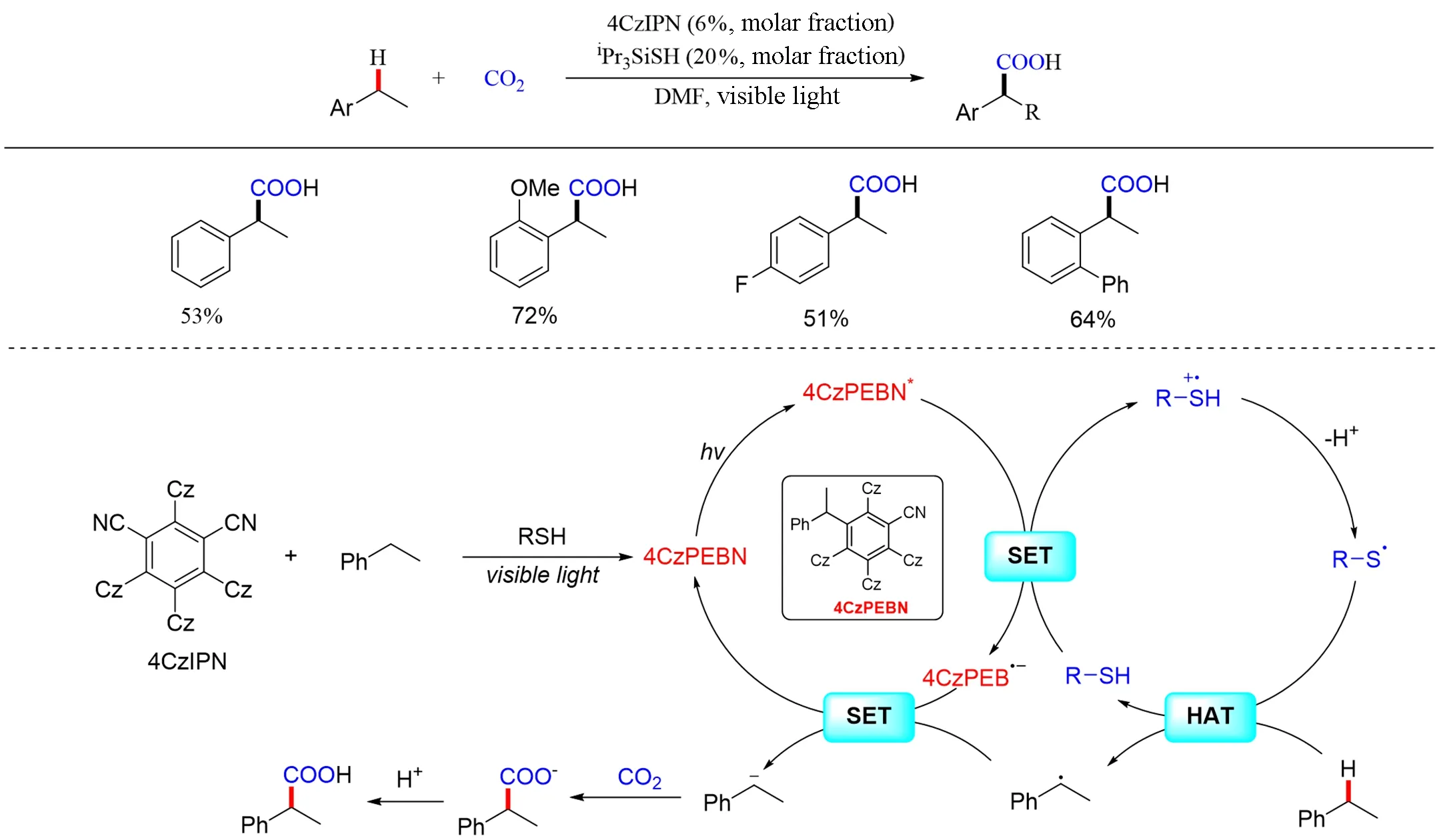
Scheme 28 Visible light photoredox⁃catalyzed carboxylation of activated sp3 C—H bonds with CO2
6 可见光促进二氧化碳参与的多组分羧基化反应
除了两组分反应,可见光催化CO2参与的多组分反应也是生成羧酸的重要方法,尤其是烯烃(或其它含不饱和键的化合物)发生的双官能团化反应. 多组分反应体系复杂,涉及如区域选择性、立体选择性等诸多问题,产物控制难度较大,但通常可以得到一系列重要的官能团化羧酸,在羧酸合成领域中具有重要意义.
6.1 二氧化碳参与的碳羧基化反应
2017年,Martin等[131]报道了可见光催化CO2参与芳基烯烃的双官能团化羧基化反应(Scheme 29).该反应用金属Ir配合物作为光敏剂,以Langlois试剂、二氟甲磺酸钠、苄基三氟硼酸钾盐或草酸单酯酸盐为自由基前体,在温和的氧化还原中性条件下实现了CO2参与芳烯烃的羧基化反应. 作者通过机理实验推测,光照下光敏剂Ir(Ⅲ)形成激发态的光敏剂Ir(Ⅲ)*,激发态的光敏剂Ir(Ⅲ)*与自由基前体发生单电子转移生成Ir(Ⅱ)和碳自由基物种,随后对芳基烯烃进行自由基加成得到苄基自由基物种,进而被还原态的光敏剂Ir(Ⅱ)单电子还原为苄基碳负离子并与CO2反应得到羧基化产物(Scheme 29). 整个反应条件温和,不需要额外加入有机金属还原剂、空气敏感的有机金属试剂及特殊的配体,为实现碳羧基化反应提供了新的策略,是对经典催化羧化反应的补充,具有广阔的应用前景.
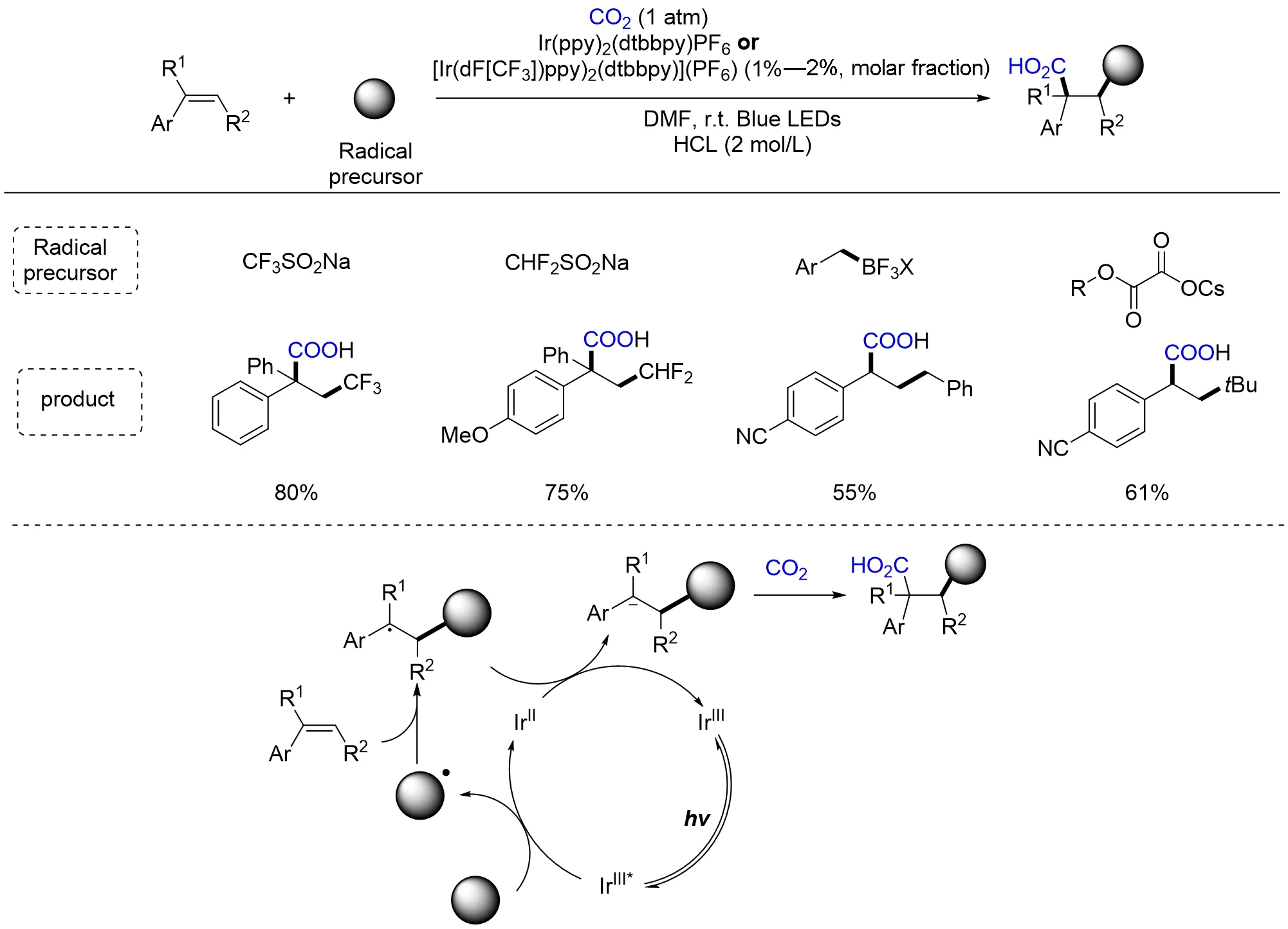
Scheme 29 Visible light photoredox⁃catalyzedintermolecular dicarbofunctionalization of styrenes with CO2 and radical precursors
2020年,Li等[132]首次以卤代物为自由基前体,实现了光催化CO2参与的烯烃碳羧基化反应,为一系列氢化肉桂酸及其衍生物的合成提供了一种高效的合成策略(Scheme 30). 该策略采用廉价易得的甲酸盐作为最终还原剂,多种易得的芳基卤代物、烷基卤代物可以作为自由基前体参与反应. 机理研究表明,体系通过HAT过程攫取甲酸盐的酰基氢形成还原性较强的CO2自由基阴离子,通过单电子还原有机卤代物得到相应的碳自由基,进而进攻烯烃,再经过单电子还原得到苄基阴离子,从而可以亲核进攻二氧化碳得到最终目标产物. 该工作不仅是可见光催化还原羧基化反应的经典例子,而且是利用CO2自由基阴离子作为还原剂实现可见光催化的重要突破. 除了有机卤代物,2021年,Sun等[133]利用亲电性的肟酯作为自由基前体,实现了可见光催化烯烃的碳羧基化反应,而且通过改变不同的光催化剂可以调控不同肟酯的转化反应(Scheme 31).
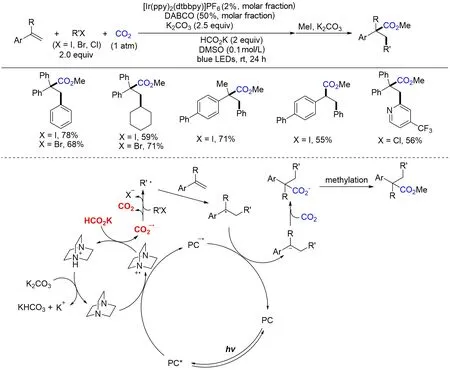
Scheme 30 Visible light photoredox⁃catalyzed arylcarboxylation of styrenes and aryl halides with CO2

Scheme 31 Visible light photoredox⁃catalyzeddicarbofunctionalization ofstyrenes with oxime esters and CO2
值的一提的是,以上反应均需要外加光催化剂才能实现,考虑到外加光催化剂合成成本高昂的问题,如果可以在无外加光催化剂的体系下实现光促进的烯烃碳羧基化反应,可能会进一步提升此类反应的实用性. 基于此,2021年,Yu等[134]利用4-烷基二氢吡啶类化合物(alkyl-DHPs)同时作为自由基前体和光激发还原剂,实现了无外加光敏剂的烯烃烷基羧基化反应(Scheme 32). 其中,4-烷基二氢吡啶类化合物能被可见光激发,分解得到烷基自由基前体,同时可以通过单电子还原烷基自由基与烯烃的加合物为碳负离子中间体,该中间体进一步进攻CO2得到目标烷基羧酸化合物. 该类反应条件温和、体系简单,多种苯乙烯和丙烯酸酯类化合物及一级、二级、三级烷基均可用于该反应,为烯烃的烷基羧基化反应提供了新思路.
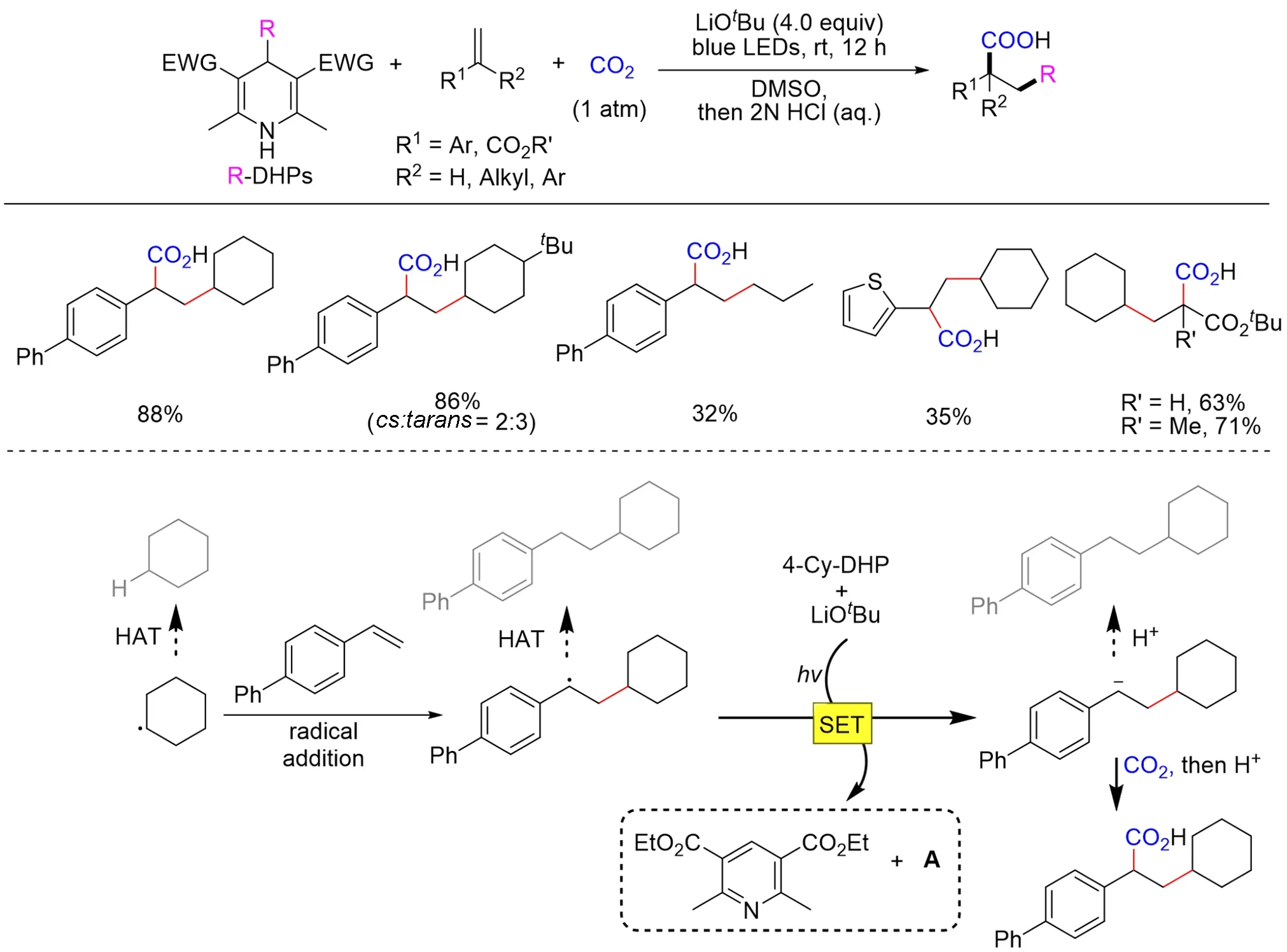
Scheme 32 Visible⁃light⁃driven external⁃photocatalyst⁃free alkylative carboxylation of alkenes with CO2
可以看出,三级胺是一个良好的电子给体,经常被用于可见光催化的还原羧酸化反应,而自身被氧化为自由基阳离子. 通常情况下,生成的该阳离子会作为副产物被忽视. 2020年,Xi等[135]利用三级胺作为α-氨基碳自由基前体和电子给体的双功能试剂,实现了可见光催化CO2参与芳基烯烃的碳羧基化反应,得到了一系列的氨基羧酸类化合物. 其中,含吸电子基团的芳基烯烃反应情况较好. 机理研究推测如下:激发态光敏剂4CZIPN被胺类化合物还原猝灭得到还原态光敏剂和胺的自由基阳离子,在碱的存在下攫取另一分子胺上的α-H形成α-烷基自由基,进而与烯烃发生自由基加成形成自由基苄基物种,该物种被还原态光敏剂还原为碳负离子,并进攻二氧化碳得到目标羧酸. 体系中锂盐的加入有利物中间体的稳定和加速单电子转移过程[Scheme 33(A)].

Scheme 33 Visible light photoredox⁃catalyzedalkylcarboxylation of styrenes and acrylates with CO2
同样是生成类似的氨基羧酸类化合物,2021年,Sun等[136]则利用简单α-氨基羧酸作为碳自由基前体,在氧化还原中性的条件下,实现了可见光催化CO2参与烯烃的碳羧基化反应. 有意思的是,当α-氨基羧酸上基团含有H时,可以实现关环反应,得到内酰胺类化合物. 作者提出如下机理:简单氨基羧酸类化合物在碱的协助下还原猝灭激发态光敏剂,脱羧得到α-烷基自由基,进攻烯烃形成新的自由基物种,该物种被还原态光敏剂还原得到碳负离,进而进攻二氧化碳得到最终目标产物;当氮上有氢时,可以脱水得到内酰胺化合物[Scheme 33(B)].
以上反应新产生的羧基均需外源二氧化碳,如果能够将脱羧反应与烯烃的双官能团化结合,把羧酸化合物作为二氧化碳源和官能团碳源双功能试剂去实现烯烃碳羧基化反应,不仅可以简化反应过程,实现绿色化学所倡导的原子经济性和步骤经济性,同时大量简单易得的羧酸类化合物可以进一步用来制备结构更为复杂的羧酸. 基于这样的理念,2021年,Yu等[137]发展了可见光催化α-氨基酸/多肽脱羧与烯烃偶合羧基化反应,高收率地获得一系列结构多样的γ-氨基酸类化合物. 反应中α-氨基酸、二肽、三肽和烷基羧酸均能与不同取代的烯烃进行反应,获得相应的γ-氨基酸化合物. 另外,该转化可以在日光下和低载量催化剂下高效进行[Scheme 33(C)].

Scheme 34 Visible light⁃driven and iron⁃promoted thiocarboxylation of styrenes and acrylates with CO2
6.2 二氧化碳参与的硫羧基化反应
2017年,Yu等[70]报道了可见光驱动的铁促进的苯乙烯和丙烯酸酯硫代羧化反应. 各种苯乙烯取代物及丙烯酸酯均有较好的反应,并以较高的收率得到β-硫代羧酸产物(Scheme 34). 重要的是,该反应羧基化发生在苯乙烯的β位,具有很好的区域选择性. 另外,该反应条件温和,具有高化学选择性和非对映选择性,产物易转化为生物活性分子及药物分子. 通过机理实验,推测机理如Scheme 34所示.首先叔丁醇钾作为碱实现硫酚的去质子化并形成铁硫复合物,可能作为重要的光致氧化催化剂或电子转移促进剂. 在可见光的照射下激发态的铁硫复合物与CO2发生单电子转移形成CO2自由基阴离子,同时形成更高价态的铁硫复合物. CO2自由基阴离子与苯乙烯发生自由基加成得到稳定的苄基自由基物种,该自由基与前边形成的高价态的铁硫复合物发生自由基-自由基偶联,形成碳硫键,进一步质子化得到最终的硫代羧酸类化合物. 该反应提供了一种新的可见光促进单电子活化CO2活化模式,为CO2高效活化以及选择性利用提供了新思路.
6.3 二氧化碳参与的膦羧基化反应
膦氧基和羧基都是非常重要的化学基团,通过烯烃官能团化将二者之一引入到化合物中目前已有许多报道. 然而,通过烯烃的双官能团化同时将两者引入到化合物中制备β-膦羧酸的例子之前尚未见报道. Yu 等[138]首次实现了可见光催化CO2参与的烯烃膦羧基化反应(Scheme 35). 在可见光驱动下,烯烃、膦氧化合物和CO2三组分可以在低量的光催化剂和适量碱的条件下反应,并以较好的收率得到一系列具有潜在生物活性的β-膦羧酸和β-膦氨基酸等化合物. 整个反应的条件简单温和,不需要额外加入有机金属还原剂或对空气敏感的有机金属试剂,为β-膦羧酸和β-膦羧酸氨基酸的合成提供了新方法. 该反应底物范围广泛,可用于芳基烯烃、丙烯酸酯、烯酰胺等一系列活化烯烃的膦羧基化. 另外,该反应可在克级规模进行,产物可进一步衍生为多种具有生物活性的化合物. 基于自由基捕获实验、自由基钟实验及氘代实验结果,作者推测该反应可能经历如下自由基历程(以烯酰胺的膦羧基化反应为例):激发态的光敏剂4CzIPN*通过单电子氧化自由基前体产生膦氧自由基物种并生成4CzIPN自由基负离子,随后该膦氧自由基物种对烯烃加成得到相对稳定的碳自由基物种,进而被还原态的4CzIPN单电子还原为碳负离子,并与CO2反应得到羧基化产物.
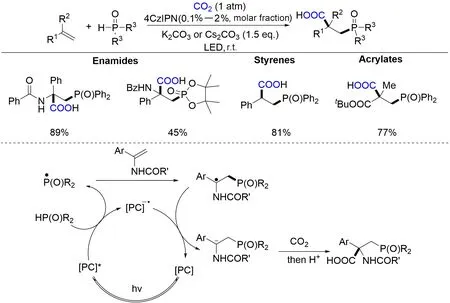
Scheme 35 Visible light⁃driven phosphonocarboxylation of alkenes with CO2
6.4 二氧化碳参与的硅羧基化反应
2018年,Wu等[139]利用光催化剂和氢原子转移催化剂的协同作用,实现了可见光催化CO2参与的烯烃硅酸羧基化反应,该体系可以利用胺类和醚类为原料,通过碳氢键活化实现烯烃的碳羧基化反应(Scheme 36). 一系列取代苯乙烯、硅烷可以与CO2反应,得到较高收率的烯烃硅羧基化产物,并且连续流技术可以实现克级规模放大. 除了缺电子的芳基烯烃外,该反应还可用于丙烯酸酯、丙烯腈及乙烯基砜等缺电子烯烃,但对于未活化的普通烯烃并不适用. 机理研究发现,激发态的4CzIPN*被3-乙酰氧基奎宁还原猝灭,得到4CzIPN自由基阴离子,同时得到3-乙酰氧基奎宁自由基阳离子中间体,进而促进硅氢键或者碳氢键的氢原子转移,生成硅基或碳基自由基和喹宁阳离子. 硅基或碳基自由基与烯烃发生自由基加在反应,得到相对稳定的碳自由基中间体,被还原态的4CzIPN还原得到碳负离子中间体,最后亲核进攻CO2得到最终产物.

Scheme 36 Visible light photoredox⁃catalyzed metal⁃free difunctionalization of alkenes with CO2 and si⁃lanes or C(sp3)—H alkanes
值的注意的是,通过合理的设计,也可以通过活化C—Si 键来实现其羧酸化反应. 2021 年,Xi等[140]首次报道了一例新型的可见光诱导的酰基硅烷与CO2的羧化反应,该反应不需要催化剂和还原剂,反应条件温和,酯化后以中等至高产率获得一系列α-酮酯. 机理研究表明,甲硅烷氧基卡宾更可能从酰基硅烷的S1状态生成,添加剂的主要作用是稳定硅氧卡宾并增强其亲核性,而生成的羧酸铯沉淀将推动反应进行. 推测反应路径如下:酰基硅烷在光照下到其单重激发态,经历1,2-甲硅烷基位移以产生单重态甲硅烷氧基卡宾中间体,随后在Cs2CO3存在下亲核进攻CO2得到羧基化产物酮酸(Scheme 37).
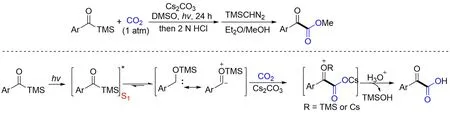
Scheme 37 Visible light⁃driven carboxylation ofacylsilanes and CO2
6.5 二氧化碳参与的烯烃远程双官能团化反应
总观以上所涉及烯烃的转化,主要局限于活化烯烃(如苯乙烯、丙烯酸酯等)的羧基化. 然而,由于普通烯烃活性比较低,利用CO2参与普通烯烃的羧基化反应存在巨大困难. 此外,二氧化碳参与的烯烃的远程官能团化,对构造结构更为多样的羧酸并将之进一步应用具有重要的意义,同时也更具有挑战性. 2021年,Yu等[141]报道了首例可见光催化CO2参与的非活化烯烃远程双官能团化羧基化反应(Scheme 38). 该反应在氧化还原中性条件下实现了CO2和三氟甲基化试剂参与非活化烯烃的远程三氟甲基化-羧基化反应,并获得了一系列含有三氟甲基的氨基酸类化合物. 该体系条件温和、底物范围广、官能团容忍性好,不仅适用于三氟甲基化-羧基化反应,也适用于二氟甲基自由基和膦氧自由基参与的烯烃双官能团化羧基化反应. 作者通过一系列控制实验和理论计算证明,该反应历经了自由基加成、1,5-氢原子转移(HAT)形成苄基自由基、单电子还原生成苄基碳负离子以及对CO2的亲核进攻等过程(Scheme 38).
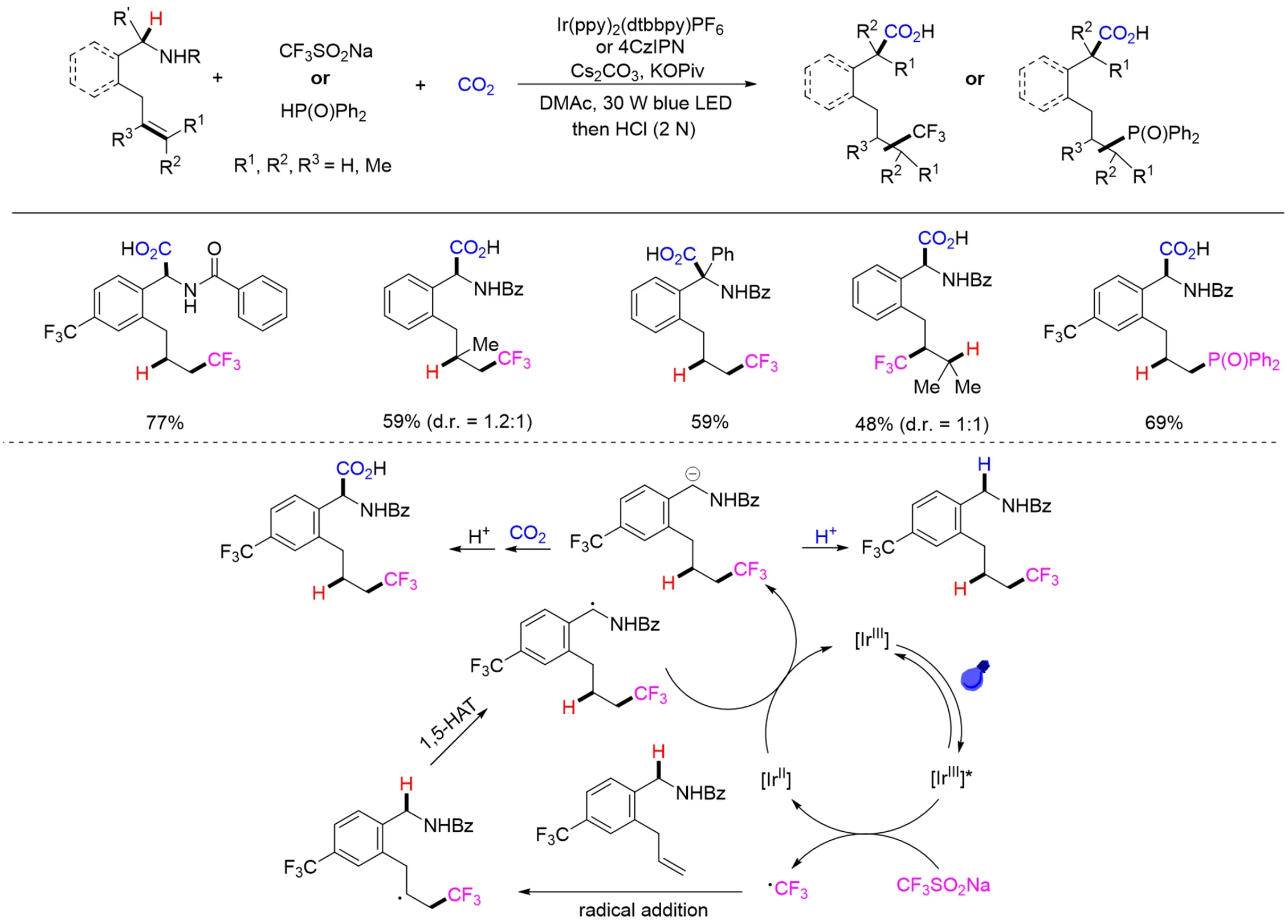
Scheme 38 Visible light photoredox⁃catalyzed remote difunctionalizing carboxylation of unactivated alkenes with CO2
7 总结与展望
随着光化学的不断发展和人们对CO2利用水平的逐步提升,光促进CO2参与的羧基化反应近年来取得了长足的进步,这些进步主要体现在:(1)反应类型的多样化、底物类型多样化发展;(2)反应所需光能由能量较高的紫外光转为可见光;(3)二氧化碳的活化模式有单电子活化和双电子活化;(4)提出多种光促进反应的催化模式.
同时这个研究领域依然有诸多具有挑战性的问题亟待解决:
(1)与二氧化碳发生反应的底物大多数是一些活性较高的反应底物,如烯烃多为苯乙烯、丙烯酸衍生物等活化烯烃,而较为惰性且更常见的普通烯烃和烷基醛酮等参与二氧化碳羧基化反应的例子虽然有报道[142],但相对较少且多数条件苛刻,同时利用二氧化碳高效合成像己二酸等大宗工业羧酸原料的研究还不成熟.
(2)在目前的反应体系下,反应规模放大尚存一定问题,虽然已出现连续流的反应方式为此类问题的解决提供了很好的思路,但是大多数反应目前还不能适用,极大地限制了光催化二氧化碳参与羧基化反应的大规模应用.
(3)由于目前应用于该领域的有机小分子光催化剂的反应活性和种类有限,导致大多数反应局限于活化底物,直接通过可见光催化二氧化碳活化和精准转化目前还鲜见报道,因此需要大力开发新型光催剂,促进该领域发展;而从目前二氧化碳利用情况来看,除以上有机光催化剂外,无机物或复合物在二氧化碳的活化中有着非常优异的表现,有望在光催化羧基反应,特别是大宗羧酸类工业原料光合成上起到重要作用.
(4)目前还没有针对性较强的、系统的理论体系来指导此类反应的开发,所以有必要加强理论指导,特别是机器学习对可见光促进二氧化碳转化的帮助[143]. 相信在各方面努力下,可见光促进CO2参与的羧基化反应将会继续蓬勃发展,成为绿色高效合成高附加值羧酸的重要方法.
猜你喜欢
中南民族大学学报(自然科学版)(2022年6期)2022-11-02
分子催化(2022年1期)2022-11-02
高等学校化学学报(2022年10期)2022-10-14
农业工程学报(2022年5期)2022-06-22
化工学报(2021年1期)2021-01-30
中南民族大学学报(自然科学版)(2020年6期)2020-12-22
中国塑料(2019年9期)2019-09-25
中国塑料(2015年10期)2015-10-14
中国洗涤用品工业(2015年9期)2015-02-28
中国塑料(2014年10期)2014-10-17
