
可见光驱动丰产金属卟啉类配合物催化的二氧化碳选择性还原反应
2022-08-06 04:37:28邱丽琪姚向阳何良年
高等学校化学学报 2022年7期
邱丽琪,姚向阳,何良年
(南开大学化学学院,元素有机化学国家重点实验室,天津 300071)
随着全球社会经济的快速发展,人类对能源的需求急剧增加. 目前,世界上85%的能源来自化石燃料(如煤、石油和天然气). 全球二氧化碳(CO2)排放量在过去30年翻了一倍,按照目前的化石燃料消耗速度,预计到2040 年将增加两倍,这将导致全球平均气温相应上升,并对环境产生深远影响[1].因此,为了有效地解决能源危机和环境问题,我国提出力争2030年前实现碳达峰,2060年前实现碳中和的目标. 研究趋势主要集中在减少化石燃料的使用,开发清洁、可再生的替代能源[2~4].
将CO2作为C1资源参与化学转化,合成具有高附加值的化学品具有重要意义及应用前景. 然而,在常温常压下CO2表现为惰性气体,其标准生成吉布斯自由能为‒394.38 kJ/mol;CO2中C=O 键的解离能为750 kJ/mol,高于许多其它化学键[如C—H(解离能430 kJ/mol)和C—C(解离能336 kJ/mol)键[5~7]],使得该分子处于最低能量状态. 在热力学上,多数CO2的化学转化是典型的吸热过程,通常需要大量的能量输入. 因此,CO2的化学转化成为极具挑战性的研究课题,关键的科学问题是温和条件下实现二氧化碳的高效活化与选择性地资源化利用[8~10]. 基于此,本课题组[11~13]率先提出CO2的捕集与利用的策略(CCU),并以此得到一系列高附加值化学品以及能源分子. 此外,我们也首次实现CO2的分级可控的2/4/6 电子还原功能化,同时构筑新的C—N 化学键[14]. 近年来,开发的人工光催化体系将CO2还原为燃料和化学品也引起了广泛关注[15~20].
光合作用是我们人类赖以生存的自然过程,植物通过叶绿素吸收光,将水和二氧化碳转化成葡萄糖、淀粉和纤维素等碳水化合物,并且释放氧气. 光合作用过程包括光反应和暗反应两部分:光反应部分吸收太阳光将水分解产生氧气,同时将光能转化为化学能,然后利用所转化的化学能在暗反应中将CO2还原成为葡萄糖. 受到大自然绿色植物光系统的启发,科学家们找到了一种缓解全球变暖和能源危机的方法,在常温常压下,通过人工光合作用将CO2转化为能源小分子[如一氧化碳(CO)、甲酸(HCOOH)、甲醇(CH3OH)、甲烷(CH4)等],即将太阳能以化学能的形式储存,真正实现能量的可持续利用. 从高能量密度和燃料长期储备的角度来看,所谓的太阳能燃料是比其它方式(如太阳能电池)更具有吸引力的太阳能存储方法[21].
由于CO2在热力学上的低能态,将CO2单电子还原为自由基阴离子的平衡电位为‒1.90 V(vs.NHE)(表1),这是一个极难发生的反应,其高能量的原因是:CO2从直线型到弯曲型结构的转变,结构上的差异造成动力学上的限制,即CO2的单电子还原法并不被认为是CO2利用的有效方法[22,23];另外一方面,质子耦合多电子转移过程中通常比单电子还原更有利,CO2可以被还原成CO,HCOOH HCHO,CH3OH或CH4,其氧化还原电位相对较低为了实现高效的CO2的还原,需要利用催化剂来降低其反应体系的活化能. 由于活化能与电化学反应的过电位相对应,高效催化剂可能在电位接近平衡电位时发生反应. 需要注意的是,与质子形成H2的还原电位‒0.41 V(vs. NHE)]相比,CO2的两电子还原在热力学上更不利,该过程中容易伴随副产物氢气的生成,因此产物选择性的调控也是面临的难题之一. 对于CO2的6电子或8电子还原,虽然热力学上是可行的,但需要在溶液中进行多次电子转移,这种多电子转移过程在动力学上是不利的. 目前对于均相催化体系而言,还原产物主要都是两电子还原产物CO 或者HCOOH,更多电子还原产物(如CH3OH,CH4等)则鲜有报道. 因此,发展可见光催化体系来实现高效、高选择性的CO2多电子还原过程是该领域长期以来的研究目标. 本文综合评述了利用均相及非均相卟啉类金属催化剂的光催化CO2选择性转化的最新进展. 首先总结了均相卟啉类金属催化体系的催化活性以及反应机理,对产物选择性调控、光催化效率的提升以及CO2还原反应机理的理解具有指导作用. 大多数均相卟啉类分子催化剂还需依赖于贵金属配合物作为光敏剂,但成本高、不利于实际应用,因此,发展易回收及重复利用的稳定催化体系成为必然趋势,还分别介绍了非均相金属卟啉基多孔有机聚合物与卟啉有机金属框架在光催化CO2还原方面的研究进展,总结分析了它们的结构与CO2转化效率和产物选择性的关系,也为后续开发高性能的廉价金属光催化CO2还原催化体系提供了新思路.
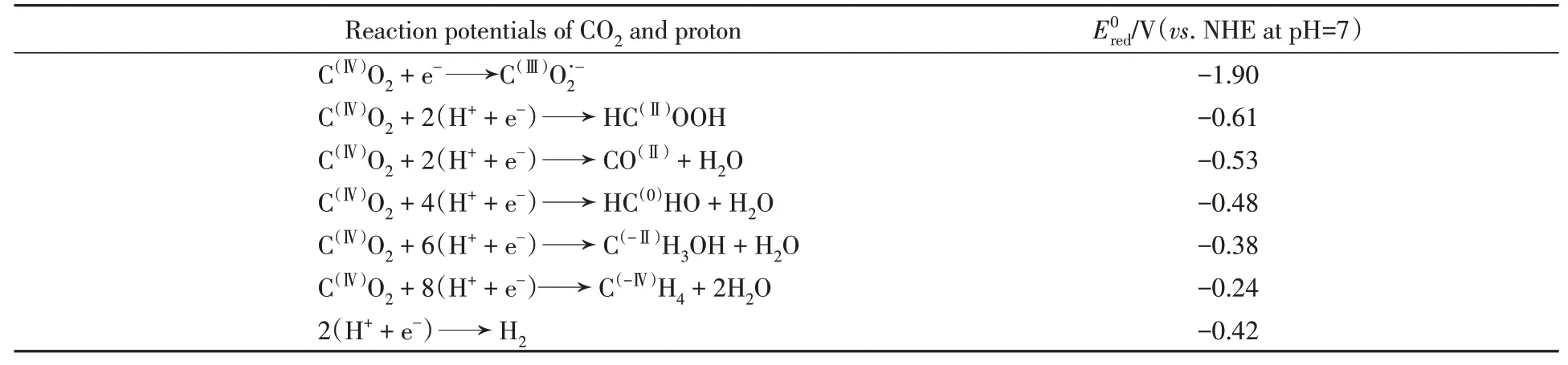
Table 1 Thermodynamic potentials of CO2 and proton reduction into various products
1 均相金属卟啉类催化剂光还原二氧化碳
1.1 卟啉铁催化剂
均相催化体系作为光还原CO2最早研究的体系,对于CO2还原的分子机理研究、新型催化剂的设计以及体系优化方面具有重要的指导意义. 金属卟啉类化合物广泛存在于自然界的生命体中,对生命活动起到重要作用,如叶绿素是卟啉镁配合物、血红素是铁卟啉配合物. 受自然界光合作用的启发,将金属卟啉类化合物用于光催化还原CO2引起了广泛的研究兴趣. 通常,选择比金属卟啉的最低未占分子轨道(LUMO)能级更负的光敏剂,可以诱导光敏剂到催化中心的电子转移,促进卟啉金属活性位点的CO2还原. 早在1999年,Neta 课题组[24]发现在铁卟啉(FeTPP)体系中添加光敏剂三联苯(TP),可以显著提高紫外光照射下CO2的转化效率,TP/TP•‒的还原电位[‒2.45 V(vs. SCE)]比FeITPP/Fe0TPP的还原电位[‒1.64 V(vs. SCE)]更低. 因此,TP可将FeITPP还原为Fe0TPP,促使CO2还原为CO. 但TP仅在紫外光驱动下才能有效地将电子传输至铁卟啉中心,从而限制了其在可见光区域的应用.
由催化剂和光敏剂组成的分子催化体系广泛应用于光催化CO2反应中,可见光响应光敏剂不仅可以为金属卟啉提供光电子以还原CO2,而且可以保护卟啉免受光解. 2014年,Bonin课题组[25]发展了一种利用9-氰蒽(9CNA)作为光敏剂和三乙胺(TEA)作为电子牺牲试剂的可见光驱动的铁卟啉衍生物CO2还原体系. 如图1(A)所示,当TEA为0.36 mol/L时[图1(A)中实心符号],45 h后CO的转化数(TONCO)达到约40. 为了限制TEA 中质子化的竞争反应,将TEA 浓度降低到50 mmol/L[图1(A)中空心符号]时,CO的生成速率依然保持线性增长,超过50 h没有观察到催化剂或光敏剂失活,TONCO为60,其选择性为100%. 当三(2-苯基吡啶)合铱[Ir(ppy)3]作为光敏剂时,CO 的选择性为93%,TONCO值高达140. 氢气的产生可能是TEA 的质子化与铁氢化物中间体的竞争反应. 在光催化系统中,光激发的9CNA 诱导了连续的两步光电子转移过程,以将Fe(III)还原为Fe(II),再还原为催化活性的Fe(0)卟啉. 随后形成了由内部氢键稳定的Fe(0)卟啉-CO2加合物,其为活化的CO2提供了两个电子,从而将CO2还原为CO[图1(B)]. 这些结果表明,将卟啉铁催化剂和廉价有机光敏剂组合,为开发高稳定性的光催化体系的研究提供了一种新的思路.

Fig.1 CO2 reduction system of iron porphyrin derivative with 9CNA as photosensitizer and TEA as sacrificial reagent(A),proposed mechanism for the photosensitized catalytic reduction of CO2 to CO(B)[25]
在已报道的分子光催化剂中,大多数催化剂主要产生CO或HCOOH,而用于获得多电子还原产物(如碳氢化合物)的催化剂仍然很少见. 2017年,Robert课题组[26]使用了带三甲基胺取代基的四苯基铁卟啉配合物(Fe-p-TMA)作为催化剂,TEA作为牺牲电子供体,用于可见光(波长λ>420 nm)驱动的CO2光还原(图2). 在室温下对含有2µmol/L Fe-p-TMA 的乙腈的1.013×105Pa CO2饱和溶液进行47 h的照射,选择性地将CO2还原为CO,TONCO为33. 未观察到副产物,CO 的生成量随时间的线性变化表明,催化系统具有良好的稳定性. 当在反应体系中额外加入0.2 mmol/L 光敏剂Ir(ppy)3,产物不仅含有CO,还能得到10%的H2和12%的CH4. 此外,0.1 mol/L 三氟乙醇的存在,使H2和CH4的选择性(TON)分别增加到19%(73)和18%(66),可能是因为三氟乙醇促进了C—O键断裂. 空白实验证实,在没有光敏剂、CO2、催化剂、电子供体或光的情况下不会形成CH4. 在12CO2或13CO2气氛下进行的同位素标记实验中,气相色谱/质谱分析分别确定为反应产物12CH4(m/z=16)或13CH4(m/z=17),确认CH4来源于CO2还原. 延长光照时间至102 h,会增加光催化产物的产量,CO,CH4和H2的TON(选择性)分别高达367(78%),79(17%)和26(5%). 该体系在光照后仍表现出稳定的紫外-可见吸收光谱,没有光敏剂Ir(ppy)3或催化剂Fe-p-TMA降解的证据,进一步说明了催化系统的稳定性. 机理研究表明,CH4出现仅在大量CO 形成后开始,这表明CO 是CH4形成过程中的中间产物. 激发态Ir 光敏剂的氧化还原电位E0[Ir(ppy)3+/Ir(ppy)3*]约为‒1.73 V(vs.SCE),这比与铁卟啉相关的3对氧化还原对(FeIII/FeII,FeII/FeI和FeI/Fe0)更负. 起始的FeIII卟啉用3个电子还原为具有催化活性的Fe0物种. Fe0物种还原CO2,FeI通过激发光敏剂的电子转移再生. 生成的CO与FeII结合,并通过FeICHO中间体用总共6个电子(从激发光敏剂转移)和6个质子进一步还原以生成CH4. 该FeICHO中间体可通过三甲基胺基团的正电荷和结合到金属的CHO物种上的部分负电荷之间的库仑相互作用而稳定. 这一认识将有助于开发更高效的催化系统,在可见光驱动下,利用丰产金属铁基配合物,将CO2先还原为CO,然后再还原为CH4.
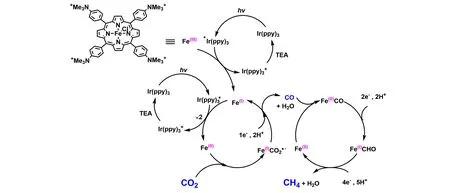
Fig.2 Proposed mechanism for CO2 reduction to CH4 by Fe⁃p⁃TMA
鉴于Ir(ppy3)作为光敏剂,Fe-p-TMA 作为催化剂已成功地用于将CO2光化学还原为CH4. 5,10-二(2-萘基)-5,10-二氢吩嗪(Phen1)和3,7-二(4-联苯)-萘-10-吩嗪(Phen2)的激发三线态还原电位分别为E0(Phen1•+/3Phen1*)=‒2.09 V(vs. Fc+/Fc),E0(Phen2•+/3Phen2*)=‒2.20 V(vs. Fc+/Fc). 其还原电位与Ir(ppy3)的E0[Ir(IV)/Ir(III)*]=‒2.13 V(vs. Fc+/Fc)接近,基于这些性质,光敏剂Phen1和Phen2在热力学上将CO2还原为CH4是可行的. 2018 年,Robert 课题组[27]利用Phen1 和Phen2 作为光敏剂,构建了Fe-p-TMA/Phen1(Phen2)体系[图3(A)],考察其对CO2转化率的影响. Fe-p-TMA/Phen2 比Fe-p-TMA/Phen1 具有更高的光催化活性,在可见光照射102 h 下,CO2饱和Fe-p-TMA/Phen2 体系的TONCO值可达140,TONCH4值可达29[图3(B)蓝色实心]. 即使经过长达4 d 的长时间辐照,催化系统仍能连续产生CH4. 以CO为原料,同一反应体系可生成CH4,选择性为85%,TONCH4值为80[图3(B)蓝色空心]. 有机光敏剂的氧化还原性质和质子源的酸性,被证明在驱动8e‒/8H+过程中发挥关键作用. 相反,在相同条件下使用Phen1 作为光敏剂仅检测到H2的产生,这可能归因于相对更正的还原电位[E0(Phen1•+/Phen1*)=−2.09 V(vs. Fc+/Fc)]. 其次,与Phen1(三重态量子产率2%和三重态寿命4.3µs)相比,Phen2具有更高的三重态量子产率(90%)和更长的三重态寿命(480µs). 值得注意的是,在相似的反应条件下,Phen2的效率明显高于Ir(ppy)3,生成的CH4的量提升约为2倍. 这为光化学催化系统开辟了新方向,可将CO2的多电子还原与水、生物质或有机化合物的氧化相耦合,可以持续制备太阳能燃料.

Fig.3 Visible⁃light⁃driven conversion of CO2 to CH4 with Phen2 and Fe⁃p⁃TMA catalyst(A),CO(black squares),H2(red circles)and CH4(blue diamonds)generation with time upon visible light irradiation(λ>435 nm) of a CO2⁃saturated(filled symbols) or CO⁃saturated(open symbols) DMF solution containing 10 μmol/L Fe⁃p⁃TMA, 1 mmol/L Phen2, 0.1 mol/L TEA and 0.1 mol/L TFE(B)[27]
1.2 卟啉钴催化剂
2019年,Sakai 课题组[28]使用水溶性苯磺酸根取代的钴卟啉(CoTPPS)作为催化剂,并使用三联吡啶氯化钌[Ru(bpy)3]Cl2作为光敏剂来光催化还原CO2. 在λ>400 nm 的光照射下,在含有CoTPPS、[Ru(bpy)3]Cl2与抗坏血酸钠(AscHNa)的CO2饱和碳酸氢盐或磷酸盐缓冲溶液中,CO的TON和转化频率(TOF)分别达到926 和456 h‒1,选择性高达82%. 有趣的是,进一步向反应混合物添加初始量的[Ru(bpy)3]2+时,会以相同的反应速率重新启动光催化系统,TON和TOF分别高达4000和2400 h‒1. 而CoTPPS或AscHNa的重新调整并未恢复初始的反应速率. 光稳定性实验也证明,[Ru(bpy)3]2+的降解是光催化失活的主要原因.[Ru(bpy)3]2+的还原电位[‒1.49 V(vs. SCE)]比CoIITPPS/[CoITPPS]‒[‒0.92 V(vs. SCE)]和[CoITPPS]‒/[CoI(TPPS•‒)]2‒[‒1.26 V(vs. SCE)]的更负. 因此,[Ru(bpy)3]2+在光照下产生电子,可将CoIITPPS 还原为[CoITPPS]‒或[CoI(TPPS•‒)]2‒. 密度泛函理论(DFT)的计算表明,CO2与[CoI(TPPS•‒)]2‒的结合是放热过程,具有较低的活化势垒,从而形成[CoI(TPPS•‒)(CO2)]2‒,最终质子化导致C=O键断裂,实现CO2到CO的转化(图4). 这项研究成功地证明了CoTPPS不仅高效地实现了水相介质中的CO2光还原,还能保持对CO的高选择性.
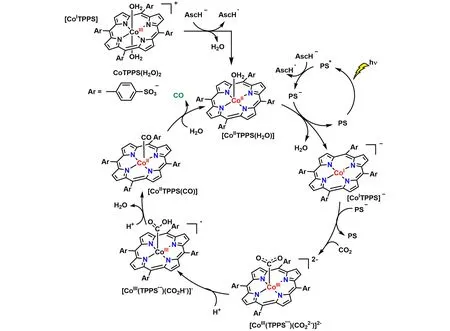
Fig.4 Proposed mechanism for the CO2⁃to⁃CO photoconversion in the [Ru(bpy)3]2+/ascorbate/CoTPPS system in aqueous media[28]
随后,在不使用贵金属的情况下,该课题组[29]开发了一种高活性和选择性的分子光催化系统,用于在纯水介质中将CO2转化为CO. 铜(Ⅰ)基水溶性光敏剂(CuPS)即使在水介质中也能保持其高发光并和长寿命的激发态. 如图5 所示,他们将4 个甲基吡啶或苯磺酸根共价取代于钴卟啉骨架上,制备了2个类似的钴卟啉衍生物(CoTMPyP或CoTPPS),进一步探索了不同芳基取代卟啉钴对CO2光还原的影响. 而CoTPPS/CuPS体系进行光驱动时表现出同样高的催化性能(TONCO=1085,CO选择性为90%). 此外,CoTMPyP与CuPS结合使用时,TONCO高达2680,表现出迄今为止的优异催化性能. 但其对CO的选择性从90%降至77%. 通过机理分析,对于CoTPPS,仅连续的单电子还原过程会在CoII位发生,以形成Co(I)活性位点并与CO2分子结合. 但是,对于CoTMPyP,在甲基吡啶受体和CoII离子处发生多电子注入以形成CoIL3‒(L=TMPyP),并有利于CO2结合形成CoIII(CO2)L(3+n)‒中间体. 因此,多电子可充电特性使最终的CO释放步骤能够通过分子内电子转移进行,这解释了CoTMPyP的高活性.
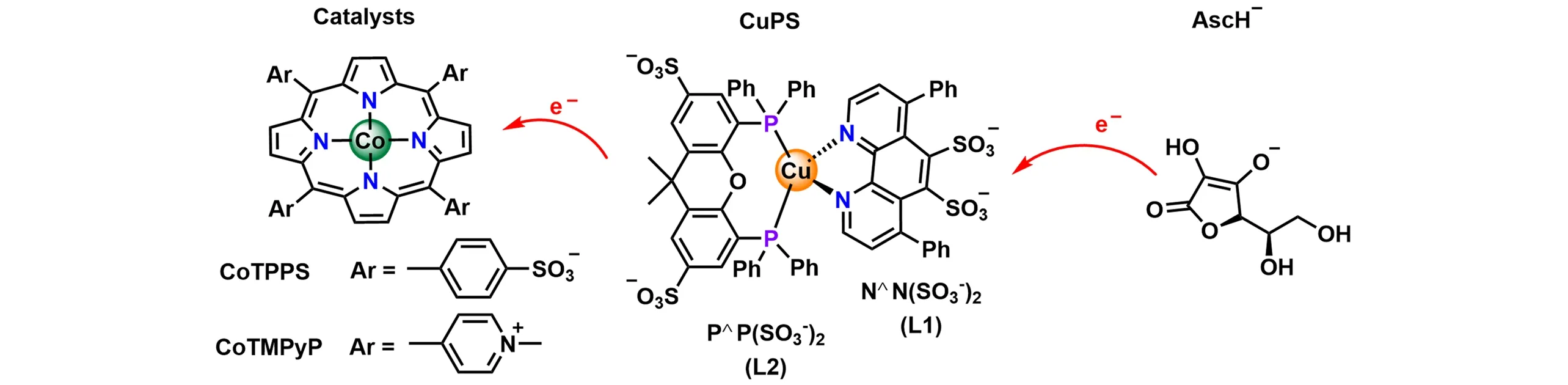
Fig.5 Photochemical CO2 reduction driven by water⁃soluble copper(I)photosensitizer with the cobalt porphyrin[29]
2 非均相金属卟啉类催化剂光还原二氧化碳
2.1 金属卟啉基多孔有机聚合物
2.1.1 无定形金属卟啉基多孔有机聚合物 尽管均相金属卟啉类催化剂在CO2还原中表现出高效率和优异的选择性,但是难以分离,因而可循环效率较低;而催化的分离与循环问题恰好是非均相催化的优势. 多孔有机聚合物作为一类新型有机半导体,具有可设计性强、比表面积大、稳定性好等优点. 卟啉材料作为非均相催化剂用于光还原CO2,因具有促进电荷转移、取代基效应等特点而受到广泛关注.传统的合成气生产方法主要是水煤气转换反应(WGSR),伴有能量消耗和大量的CO2排放[30]. 因此,利用可见光诱导CO2光还原制合成气(Syngas)是为实验室和制药工业提供合成气代用品的一种有吸引力的方法.
本课题组[31]发展了一种易于操作的方法来调控CO2还原反应和析氢反应(HER)活性制备合成气,而不是完全抑制氢气的产生. 与传统的引入多组分催化位点或调解溶剂中有机溶剂/水含量的方法不同,我们设计并制备了铁卟啉基多孔有机聚合物半导体[POPn-Fe,n=1(苯基为连接单元)或2(联苯基为连接单元)][图6(A)],CO/H2的收率最高可达3043/3753µmol∙g‒1∙h‒1,比例可控制在1∶1~1∶2 之间[图6(B)]. 其中非配位的卟啉和铁卟啉位点分别促进了H2和CO的形成,铁物种也降低了催化剂的阻抗,从而提高了电荷迁移率,更利于将电子传递给CO2或质子. 联苯连接单元加速了光生电子和空穴的分离,由于低聚苯的高HOMO能级,在聚合物骨架中苯单元替代联苯单元,导致了POP1-Fe整体的LUMO 能级向真空移动,过高的还原电位不利于POP1-Fe 从光敏剂中夺取电子,因此,POP2-Fe 比POP1-Fe具有更高的催化活性,对合成气形成具有良好的CO选择性. 另一方面,POP2-Fe不仅提供了丰富的表面活性位,荧光猝灭实验和荧光寿命也表明,其也促进了空穴电荷分离和电子转移. 这可能是由于交联的POP框架上形成了高度离域的π键,从而有利于从激发态的光敏剂中捕获电子. 该研究在引入不同催化位点以充分利用竞争性HER、改变连接单元以调控催化活性及加速电荷转移以促进载流子分离等方面,为进一步通过研究太阳能驱动的CO2转化为合成气提供了启示.
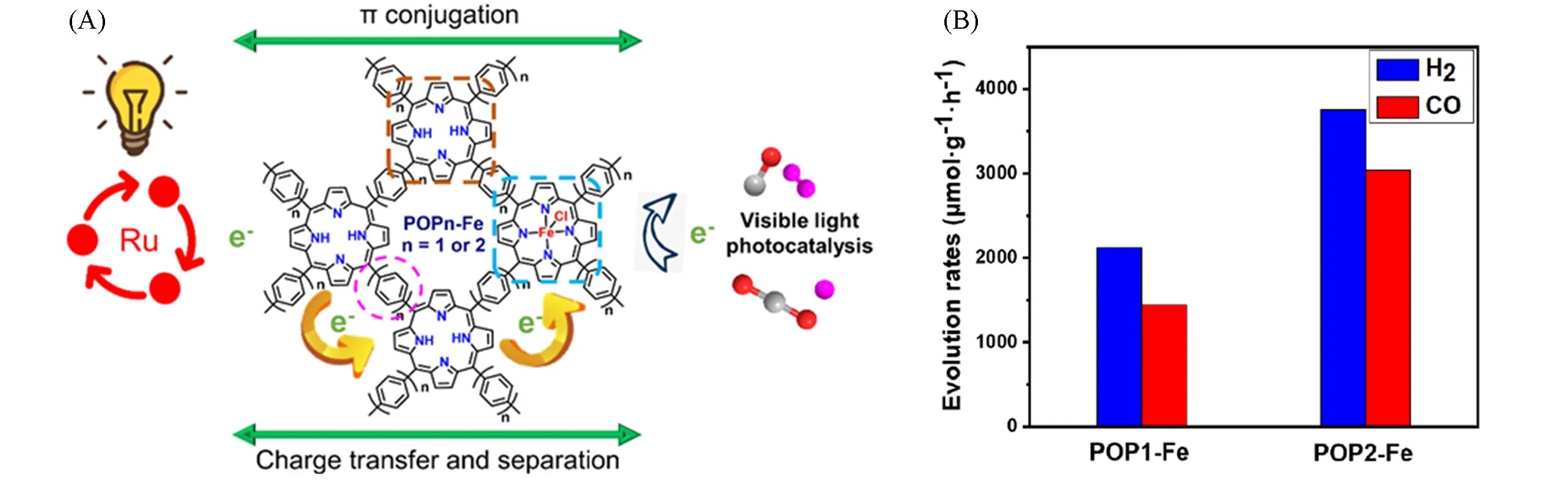
Fig.6 Ferric porphyrin⁃based porous organic polymers for CO2 photocatalytic reduction to syngas with selectivity control(A),photocatalytic syngas reduction performance of POPn⁃Fe(B)[31]
2.1.2 结晶性金属卟啉基共价有机框架 共价有机框架(COF)是一种新兴的晶体多孔材料,由牢固的共价键和刚性有机结构单元组成. 该类材料具有较高的比表面积、规整有序的孔结构[32]. 席夫碱反应是形成COF的典型缩合反应,可以将金属卟啉整合到框架中,以构建用于光催化CO2转化的高度结晶性与孔道丰富的COF 材料. 利用太阳能将CO2转化为燃料,并以H2O为牺牲剂,是光合作用中一个具有挑战性的研究领域. 在此基础上,2019年,兰亚乾课题组[33]合成了一系列四硫富瓦烯卟啉共价有机骨架TTCOF-M(M=Zn,Cu,Ni),并将其作为光催化剂,在不添加光敏剂、牺牲剂和贵金属共催化剂的情况下,用于CO2还原和H2O 的氧化反应[图7(A)]. 其中,TTCOF-Zn 的价带比O2/H2O 的氧化电位更正,而导带则比CO2/CO的还原电位更负. 在可见光(420 nm<λ<800 nm)照射60 h下,TTCOF-Zn可以实现完整的人工光合作用,CO和O2的总生成量分别为12.33和6.17µmol[图7(B)]. 机理研究表明,通过共价键合四硫富瓦烯能有效地将光生电子转移到卟啉单元上,产生分离的电子和空穴分别进行CO2还原和H2O氧化. 该报道以H2O为电子供体,设计合理的晶体COF体系用于CO2的选择性光还原反应,为理解非均相光催化剂的结构-功能关系提供了更直接、更清晰的晶体证据,也为用于人工光合作用晶体光催化剂的设计提供了新的视角.
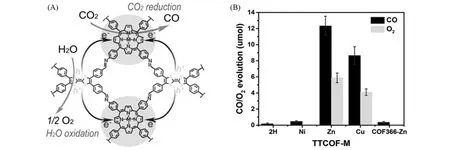
Fig.7 TTCOF⁃M catalyst for efficient CO2 photoreduction with H2O(A), CO2 reduction performance of TTCOF⁃M and COF366⁃Zn(B)[33]
2020年,王瑞虎课题组[34]利用四(4-氨基苯基)卟啉和4,4′,4″,4‴(乙烷-1,1,2,2-四基)四苯甲醛(TPE)合成了一种基于卟啉的COF(MP-TPE-COF,M=H2,Co和Ni),其能选择性光催化CO2转化为CO[图8(A)]. NiP-TPE-COF 对CO 生成具有93%的高选择性,生成速率为525µmol∙g‒1∙h‒1. 而对于CoPTPE-COF,CO 的生成速率高达2414µmol∙g‒1∙h‒1,但是CO 的选择性仅有61%[图8(B)]. DFT 计算表明,NiPTPE-COF 在从CO2-MP-TPE-COF 到COOH-MP-TPECOF 中间体的质子化过程中具有更高的吉布斯自由能(ΔG)[图8(C)],表明NiP-TPE-COF 中H2生成之前就具备CO2转化为CO 的趋势,因此导致NiP-TPE-COF中的高CO选择性. Co物种与CO2结合所需能垒比NiPTPE-COF更低,促使CoP-TPE-COF具有较高的光催化还原性能.
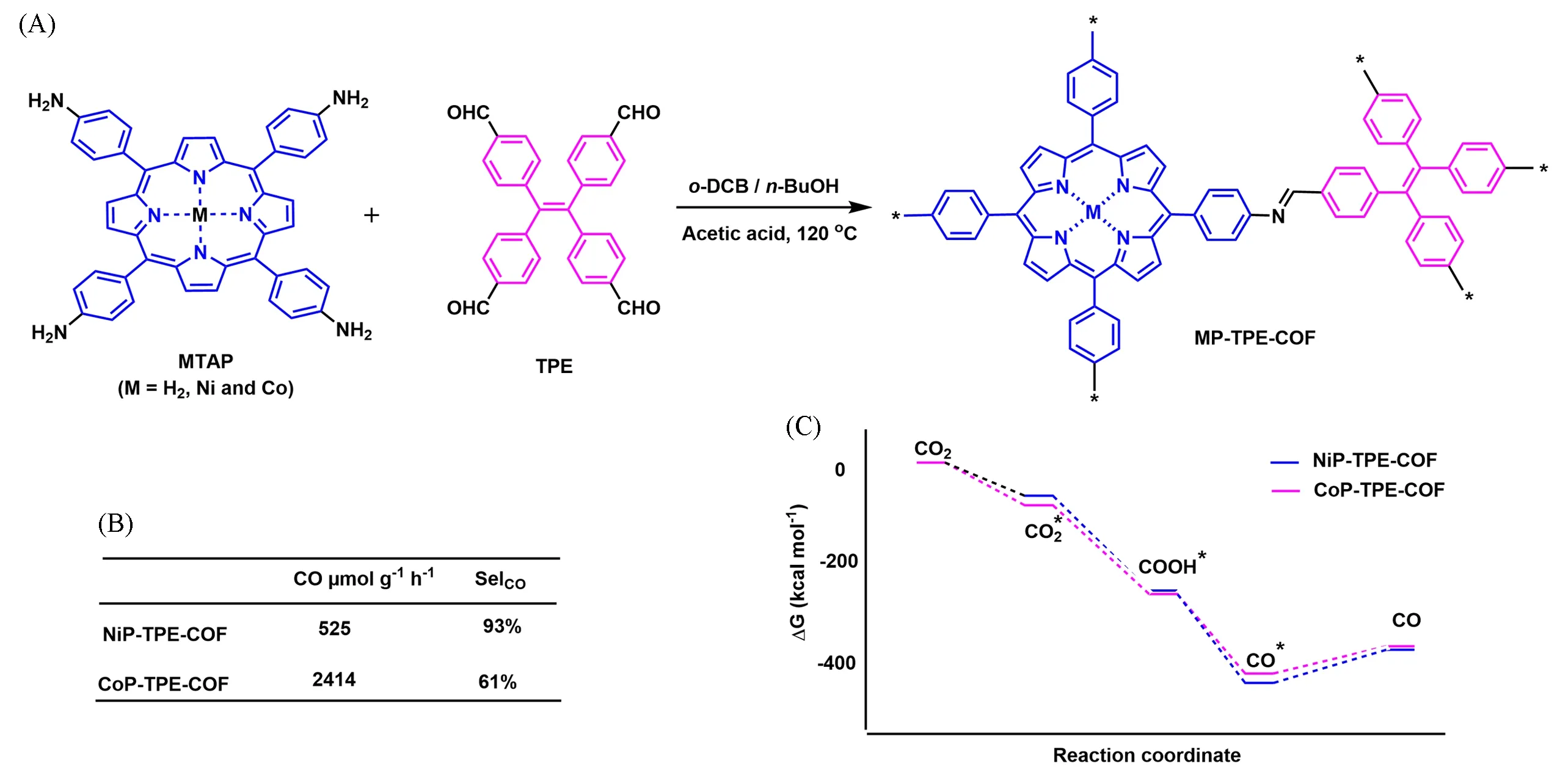
Fig.8 Schematic depiction for the synthesis of MP⁃TPE⁃COF(A),CO2 photoreduction over NiP⁃TPE⁃COF and CoP⁃TPE⁃COF(B),DFT⁃calculated ΔG profiles for the CO2⁃to CO conversion(C)
2.2 卟啉有机金属框架
由有机配体和金属离子/团簇组装而成的金属有机骨架(MOF),在光催化还原CO2中引起了研究人员极大的兴趣. 金属离子可以很容易地结合到卟啉单元中以产生配位不饱和的金属位点. MOFs中分散均匀的多孔性可以确保每个活性位都容易与CO2分子接触[35]. 因此,这些光催化系统不仅可实现分子CO2的活化和转化,也能从分子水平对其结构进行精确调控. 锆(Zr)节点簇由于其相对较强的稳定性,被广泛用于构建基于卟啉的MOF(PMOF)并用于CO2光还原[36]. 卟啉可以收集光并作为电子给体,将光生电子转移至Zr节点簇,从而将ZrIV还原为ZrIII,以进行CO2的吸附和光还原.
2015 年,江海龙课题组[37]通过四(4-羧基苯基)卟啉(H2TCPP)和四氯化锆(ZrCl4)的自组装形成稳定的基于Zr的介孔PMOF(PCN-222)[图9(A)]. PCN-222的导带电位比CO2形成HCOOH的还原电位更负[‒0.28 V(vs. NHE)],因此在热力学上能够驱动CO2还原为HCOOH. 在可见光照射下,PCN-222在MeCN/三乙醇胺(TEOA)溶液中实现CO2到HCOO−的转化,HCOO−生成量在10 h 内可达30 µmol[图9(B)]. PCN-222中的强CO2吸附能力(298 K下的吸收量为35 cm3/g)可使其能够更好地与CO2在乙腈溶液中相互作用,从而有利于其光催化高效转化. 瞬态吸收和光致发光光谱结果证明,PCN-222中的长寿命电荷分离态能有效地抑制光生电子-空穴对复合,因此促使了CO2光还原的高效率. PCN-222中的H2TCPP可有效吸收可见光,并将光生电子转移到Zr氧簇中,ZrIV被还原成ZrIII,以在PCN-222上进行CO2光还原. 这项研究突出了将卟啉引入MOF中以进行CO2光还原的优势,为深入了解PMOF 中的电子转移机理提供了新的思路.
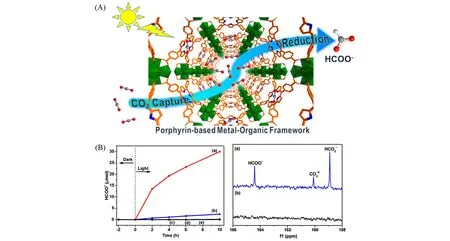
Fig.9 A porphyrin⁃involved MOF(PCN⁃222) promoted photocatalytic CO2 reduction(A), amount of HCOO−produced as a function of the time of visible⁃light irradiation and 13C NMR spectra for the product obtained from reaction with 13CO2 or 12CO2(B)[37]
卤化铅钙钛矿量子点具有可调的可见光吸收范围、可调的能带结构以及较大的摩尔吸光系数,是一类极具吸引力的光催化材料. 提高卤化铅钙钛矿型量子点(QD)在含水体系中的稳定性,是其在人工光合作用中实际应用的关键. 2019 年,鲁统部课题组[38]将有机金属卤化物[CH3NH3PbI3(MAPbI3)]量子点封装到富含铁卟啉的金属有机骨架PCN-221(Fex)的孔隙之中,以构建可见光驱动的MAPbI3@PCN-221(Fex)(x=0~1)光还原CO2复合材料[图10(A)]. 在MOF的保护下,复合光催化剂在含水的反应体系中表现出更高的稳定性. 将MAPBI3QD 封装在PCN-221(Fex)的孔中不会改变其晶体结构,但明显增强了可见光吸收能力. 超小型MAPbI3量子点能够进入PCN-221的腔体,导致MAPbI3量子点与MOF中的铁卟啉部分紧密接触. 稳态和时间分辨光致发光实验表明,封装的MAPBI3量子点中的光生电子可以快速转移到铁催化位点,实现钙钛矿量子点和铁卟啉基MOF之间的有效电荷分离. 复合光催化剂MAPbI3@PCN-221(Fe0.2)具有极高的光催化活性,可将CO2还原为CO(34%)和CH4(66%),比相应的PCN-221(Fe0.2)高38倍[图10(B)]. 这项研究提供了一种通过顺序沉积途径合成均匀量子点封装的PMOF基质的方法.
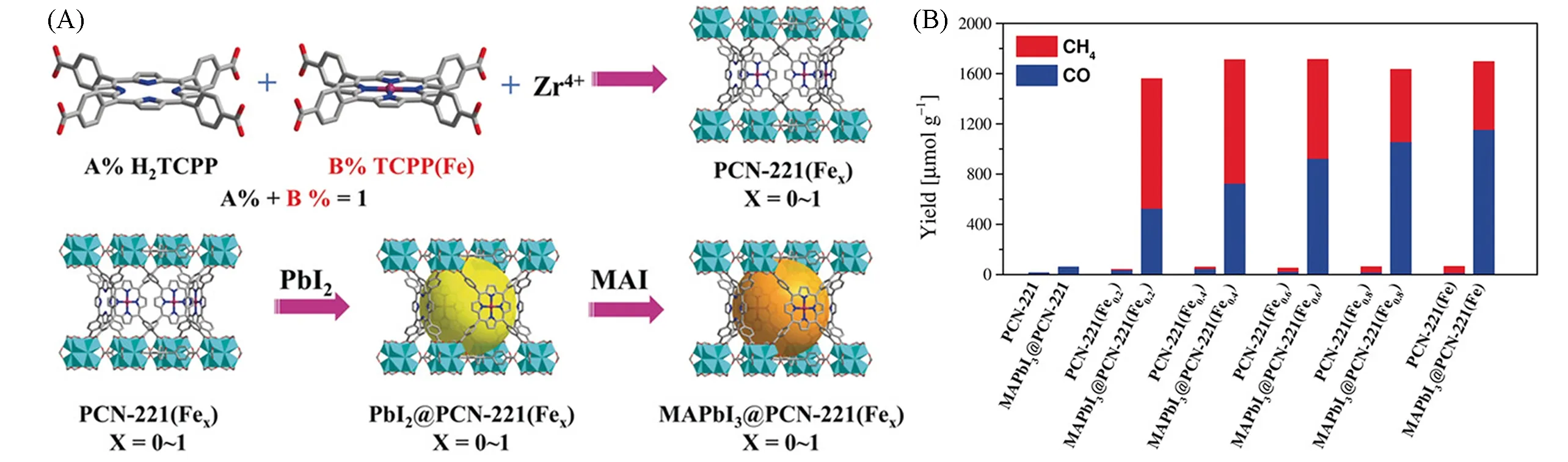
Fig.10 Encapsulating perovskite quantum dots(CH3NH3PbI3)in PCN⁃221(Fex)for efficient photocatalytic CO2 reduction(A), the yields for CO2 reduction to CH4 and CO with PCN⁃221(Fex)and MAPbI3@PCN⁃221(Fex)as photocatalysts(B)[38]
目前,将CO2高选择性地生成碳氢燃料(如CH4)是一项极具挑战性的工作. 2020 年,兰亚乾课题组[39]将强还原性多酸簇(Zn-ε-Keggin)与光敏性的四(4-羧基苯基)卟啉(H2TCPP)连接单元相结合,制备了两种稳定的多金属氧酸盐接枝金属卟啉配位框架(POMCFs:NNU-13及NNU-14). X射线单晶衍射分析表明,NNU-13 为斜方晶系空间点群,NNU-14 为单斜的C2/m空间点群[图11(A)]. 在含有NNU-13或NNU-14光催化CO2还原系统中,实现了水相中CO2到CH4的还原,高选择性高于96%[图11(B)]. 而NNU-14的CH4选择性(96.2%)略低,主要受空间构型变形影响. 值得注意的是,引入具有强还原性的Zn-ε-keggin团簇是POMCFs获得高光催化CH4生成选择性的重要来源,考虑到8个钼(MoV)原子理论上可以提供8个电子,来完成CO2到CH4转化的多电子还原过程. 多酸-卟啉配合物为光电催化机理研究提供了很好的晶体模型,研究表明,光催化过程中光驱动电子从卟啉向多酸移动,多酸表面聚集的更多电子有利于CH4的生成. 对比单金属和金属簇CO2还原催化产物的区别,明确了催化位点及机理.值得注意的是,Zn-ε-keggin团簇的强还原能力,及与卟啉单元连接单元的优异光学和电学性质,使这些化合物具有优异的光催化生成CH4的选择性.
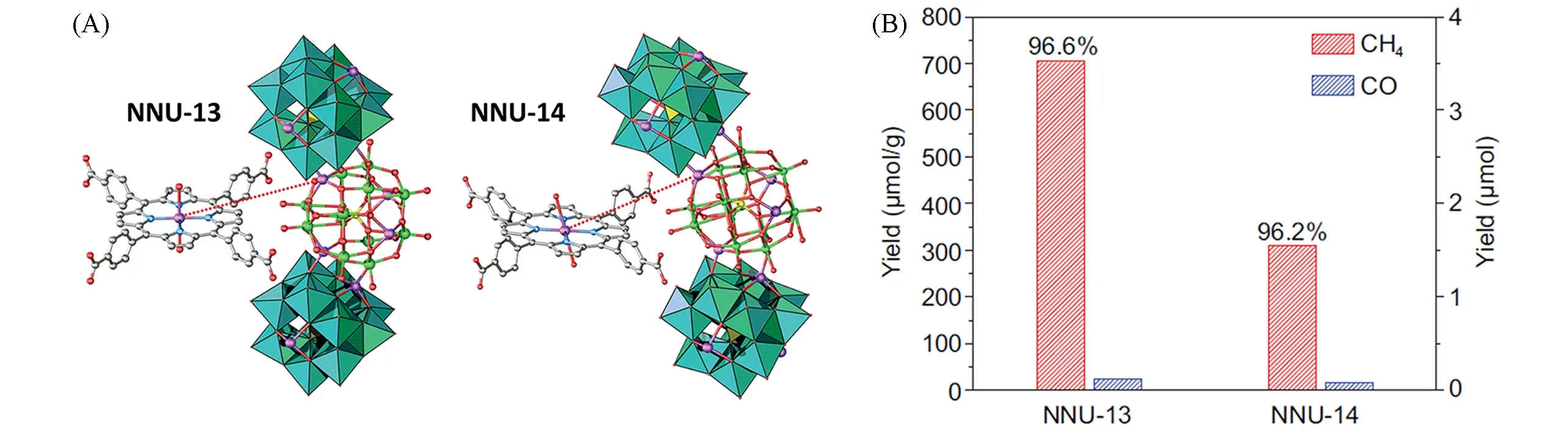
Fig.11 Polyoxometalate⁃grafted metalloporphyrin coordination frameworks for selective CO2⁃to⁃CH4 photoconversion(A), the total product yield and selectivity of gas products in the photoreduc⁃tion of CO2(B)[39]
最近,对卟啉环次甲基连接的外围基团进行修饰,可以产生具有不同结构和性质的PMOF. 曹荣课题组[40]报道了吡唑基卟啉Ni-MOF(PCN-601)(图12),其由Ni-氧代簇节点和通过吡唑基连接的Ni-卟啉单元组成. 与上述羧基卟啉MOF相比,吡唑基具有更大的π共轭,并且可以与Ni-氧代簇节点产生更高的d-π轨道重叠,从而加速配体到节点的电子转移并抑制光生电荷重组,使整个CO2还原过程具有较高的活性和稳定性. 在水蒸汽的条件下,由可见光驱动的整体气相CO2还原,CH4的生成速率达到10.1µmol∙h‒1∙g‒1,光催化效率远远超过了类似的羧基卟啉MOF和众所周知的无机Pt/CdS光催化剂. 该反应避免使用电子牺牲试剂,并在气相中进行,充分利用MOF的强气体吸收特点. 这项研究表明,MOF结构中配位球的合理设计,不仅协调了反应性和稳定性之间的矛盾,而且极大地促进了界面电荷转移,为高效MOF光催化剂的设计提供了指导.
Fig.12 Pyrazolyl porphyrinic Ni⁃MOF(PCN⁃601)for selective CO2⁃to⁃CH4 photoconversion[40]
3 总结与展望
工业革命后,化石资源的消耗导致大气中CO2浓度不断上升和全球储备能源持续减少,由此导致了温室效应和能源短缺问题. 光还原CO2能够直接利用太阳能作为驱动力,模拟光合作用来构筑新的碳循环路径,实现可再生能源驱动的CO2可持续资源化的新途径. 本文总结性地概述了多种类型的卟啉类金属配合物在均相和非均相光催化还原CO2选择性转化的相关进展,光还原CO2可选择性生成CO,HCOOH 和CH4等碳基燃料. 均相卟啉体系为研究金属活性中心和配体对调控其催化活性提供了很好的研究参考. 但均相卟啉类金属配合物也存在稳定性不高,反应过程中需要光敏剂的参与,难以应用到工业化生产等问题. 基于对均相催化机理的理解,人们也开发了非均相催化体系. 框架材料与聚合物体系中的不同催化位点和COFs的有机连接体,使卟啉骨架具有不同的拓扑、形态和孔径. 尽管如此,如何提高催化剂的活性、选择性和稳定性仍是有待突破的课题.
利用可持续太阳能,通过非均相光催化实现CO2的资源化,具有操作简单、便于连续工业过程应用、可循环利用等优点. 因此,其为产生高能电子和空穴以触发氧化还原转变提供了一条经济和环境上有吸引力的途径. 在光催化CO2转化的各种反应体系中,模拟CO2还原与H2O氧化耦合的性质是一种理想的绿色过程. 然而,H2O是一个较差的电子供体. 另一方面,O2和H2O可能接受电子与CO2还原半反应竞争,并产生一系列破坏光催化体系的活性氧物种. 此外,在中性pH下CO2在H2O中的溶解度较低,CO2光还原过程将与H2的生成存在竞争. 这些热力学和动力学限制共同导致了光催化CO2转化的低效率和低选择性. 为了解决这些问题,通常使用TEOA,TEA等牺牲剂作为电子供体来捕获空穴,以促进CO2光还原. 然而,这种策略存在不必要地浪费空穴能量、生成无价值的氧化产物,从而并增加系统成本的弊端.
针对出现的问题,以后的发展趋势可以从以下几个方面考虑:(1)在设计反应时将是否以工业化反应条件为目标;(2)设计CO2多电子还原的催化体系,实现C2+以上多碳产物的生成;(3)有机底物可以取代H2O产生增值化学品,同时注入还原当量的质子和电子,有利于CO2的活化与还原,从而提高耦合反应系统的稳定性和整体催化效率. 因此,设计和合成有效的光催化剂以实现该类反应是至关重要的. 耦合策略能有效地协同利用电子和空穴,在实现可持续的光氧化还原催化方面更具吸引力,符合绿色化学和可持续发展要求;(4)另外,为了降低成本、简化分离回收、实现光催化的工业放大,将催化剂负载或者固定在基底上似乎是必然趋势,对于非均相卟啉基材料的设计要考虑到催化位点的暴露、结构与活性的关系与大规模制备等问题;(5)已发展的大多数是双组分光催化体系,可通过分子间(光敏剂到催化中心)的电子传输来驱动CO2还原反应,并可借鉴Ishitani 课题组[41~45]的超分子催化体系,通过共价键桥连不同的廉价光敏剂与卟啉金属类催化剂来发展新型的催化体系,以促进更快速的电子转移;(6)发展双金属中心协同催化的光催化CO2还原体系,有利于CO2的活化与固定,加速反应进程,并为选择性获得多电子还原产物(如甲醇或烃类)开辟新途径.
猜你喜欢
新作文·中学作文教学研究(2022年4期)2022-08-25 02:38:48
太原科技大学学报(2020年3期)2020-06-22 01:27:26
山东化工(2019年2期)2019-02-16 12:38:10
陶瓷学报(2019年5期)2019-01-12 09:17:34
福建农林大学学报(自然科学版)(2018年5期)2018-10-11 08:05:32
三峡大学学报(自然科学版)(2017年1期)2017-03-20 15:30:23
中国资源综合利用(2016年9期)2016-01-22 08:35:22
支点(2015年11期)2015-11-16 10:25:03
应用化工(2014年7期)2014-08-09 09:20:26
中国药理学通报(2014年2期)2014-05-09 08:22:16
