
二氧化碳间接转化制化学品的研究进展
2022-08-06 04:37:22黄孝舜马海英柳淑娟王红利崔新江
高等学校化学学报 2022年7期
黄孝舜,马海英,柳淑娟,王 斌,王红利,钱 波,崔新江,石 峰
(中国科学院兰州化学物理研究所,兰州 730000)
工业革命中化石燃料的使用促进了人类现代化进程,但其无节制的使用则是导致大气中温室气体排放量剧增的最根本原因,其中CO2约占温室气体总排放量的3/4[1~4]. 从1850年至2018年,全球平均温度已经升高超过了1 ℃[5~7],而由此导致的冰川融化、海平面上升以及极端天气等已经威胁到人类和动物的生存环境[8~13]. 我国提出将提高国家自主贡献的力度,采取更加有力的政策和措施,CO2排放力争于2030年前达到峰值,争取在2060年前实现碳中和[14]. 为了实现上述战略目标,应当使用一切可能的手段进行CO2的利用和存储. 近年来出现了几种有利于CO2减排的应对手段,包括发展更高效环保的发电技术[15~19]、提高能源利用效率、发展清洁能源技术(煤制气)、碳捕集与封存技术[20~27]. 基于成本、能源资源分布和实施时间、规模效应等因素,利用CO2转化为有用的燃料和化学品,可以有效增强CO2的利用率,从而减少温室效应并提供清洁能源. CO2转化和利用主要通过化学/催化转化(化学过程)、酶转化(生物/生化过程)和技术利用(物理过程)等方法. 充分利用CO2资源不仅可以缓解温室效应,而且也可生产高附加值的含碳化学燃料和化工产品.
其中,CO2的化学转化是实现CO2大规模利用的有效途径之一[28,29],但CO2是所有燃烧过程中的最终产物之一,具有动力学惰性. 且CO2的转化在热力学上是不利的,大部分CO2化学转化反应在热力学上都是非自发的(ΔG>0)[30]. CO2的化学利用主要有两种途径:(1)CO2直接还原为低碳化学品(C为≤+2的还原态);(2)CO2转化为高碳有机化学品(C为+4或+3的氧化态). 其中,第一种反应途径存在高能耗、低活性、产品选择性不足及催化剂稳定性差等问题[31~39],而第二种途径可以在温和的条件下实现将CO2进一步转化利用为高附加值化学品[40~49]. 目前,CO2直接转化的产率普遍较低,通过CO2直接加氢高效制备高附加值化学品较为困难[50,51];相较于CO2直接转化,CO2转化为接力分子继而间接转化为高附加值的化工产品具有很大优势. 首先,接力分子中羰基的活性相对较高,易于活化,有利于催化反应的发生;其次,副产物可以回收利用,使得合成过程更加绿色高效[52~55].
在CO2间接转化中,将CO2转化为二取代碳酸酯的过程,因其原子利用率高、环境友好而备受关注[56~58]. 环碳酸酯是一种CO2衍生的二取代碳酸酯,是CO2间接转化利用的良好选择之一. 碳酸乙烯酯(Ethylene carbonate,EC)是其典型代表,其原料来源广泛,通过CO2与环氧乙烷[59]或者乙二醇[60]的反应很容易得到EC,且CO2与环氧乙烷制备EC已实现工业化,通过这一反应途径,在较温和的条件下,进一步对EC进行转化可获得高附加值化学品(如甲醇、碳酸二甲酯、乙二醇及恶唑烷酮等),展示了EC作为CO2间接转化重要中间体的应用潜力. 目前,EC的合成、酯交换、聚合以及在电池、生物医学方面的应用已有了较详细的总结[61~70],本文主要综合评述了近年来以EC为原料,与氢气,醇、胺反应的研究状况,以期为积极有效开展此方面的研究提供有益的借鉴.
1 环碳酸酯的加氢反应
甲醇是最基本的有机化工原料,自身产业链长,涉及化工、建材、能源、医药和农药等很多行业,在国民经济中具有重要地位. 目前,CO2直接加氢的转化率和选择性普遍较低[54,71,72],而通过将CO2转化为环碳酸酯,再加氢制备甲醇则是更高效的转化路线(Scheme 1).
目前,对于环碳酸酯加氢反应报道的催化剂主要集中于Ru 基均相催化剂和Cu 基多相催化剂. 如表1所示,Ru基均相催化剂普遍具有很高的催化活性,但均相催化剂分离困难,且贵金属催化剂价格昂贵,限制了其在工业生产中的应用[73~82]. 而有关EC加氢反应的多相催化剂主要集中于少数几种Cu基催化剂,其催化效率和催化剂的稳定性仍有待进一步提升.
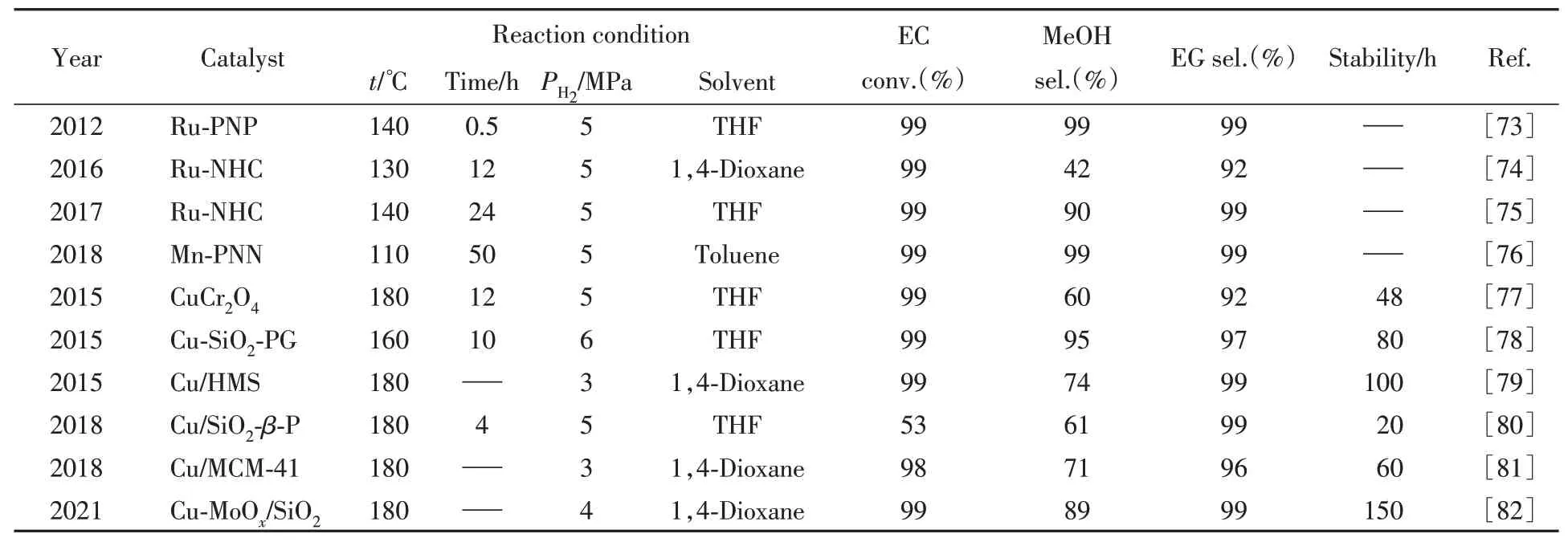
Table 1 Catalytic system of EC hydrogenation
1.1 均相催化剂
Milstein 等[83]于2011 年首先开发一种将CO2间接转化为甲醇的方法,他们使用[(PNN)Ru(CO)(H)]实现了在相对温和的条件下,从CO2到碳酸二甲酯再到甲醇的转化. 但因原料碳酸二甲酯比产品甲醇贵得多,故难以实现工业化生产. 因此,研究人员致力于开发新的反应途径和实用的催化剂体系.
2012 年,丁奎岭团队[73]采用Ru-PNP 配合物为催化剂,首次实现了从CO2到EC 再到甲醇的转化,并在EC加氢反应中表现出优异的催化活性. 该工艺为从EC同时生产两种重要的大宗化学品(甲醇和乙二醇)提供了一种可行的工艺路线. 与此同时,他们对多种有机环状碳酸酯使用该催化体系进行了加氢反应的研究,均具有良好的效果(Scheme 2).
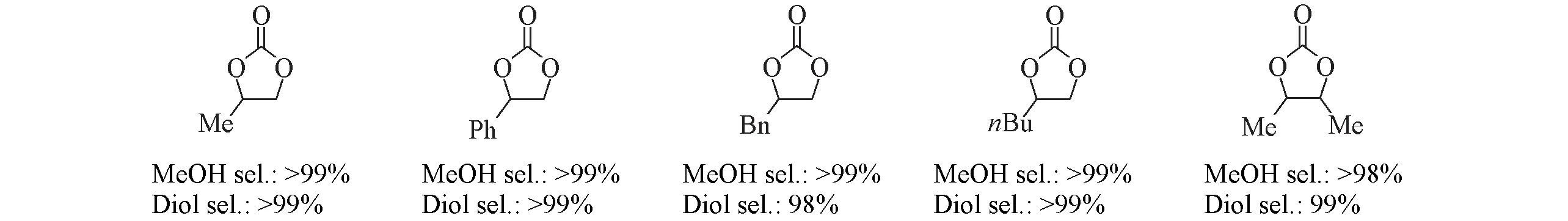
Scheme 2 Hydrogenation of various cyclic carbonates in the presence of RuII complex
高国华团队[74]研制了一种Ru-NHC配合物均相催化剂,在EC加氢反应中具有较好的性能. 其中,N-丁基取代的NHC配体和RuHCl(CO)(PPh3)3Ru前驱体所形成的Ru-NHC配合物表现出最高的催化效率,认为NHC配体和Ru前驱体原位生成Ru-NHC配合物可能是该反应的活性催化剂.
随后,涂涛团队[75]成功合成了一系列吡啶桥接的Ru-NHC 配合物,在温和的反应条件下,即可在各种环状和直链碳酸酯的氢化反应中表现出非常高的催化活性. 即使在低催化剂负载和弱碱催化量的情况下,具有不同环大小和空间位阻环状碳酸酯的各种底物也具有良好的耐受性,都可以得到较好的甲醇及其相应的二醇产率(Scheme 3).
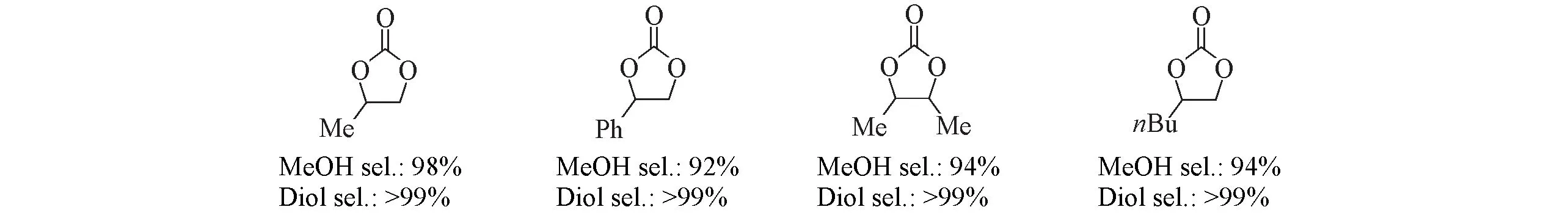
Scheme 3 Hydrogenation of various cyclic carbonates in the presence of Ru⁃NHC
此外,Milstein 团队[76]合成了一系列Mn-PNN 催化剂,在110 ℃、5 MPa H2、50 h、甲苯为溶剂条件下,EC转化率>99%,甲醇与乙二醇的收率均大于99%,且对于其它类型的环碳酸酯也有较好的活性和选择性(Scheme 4).
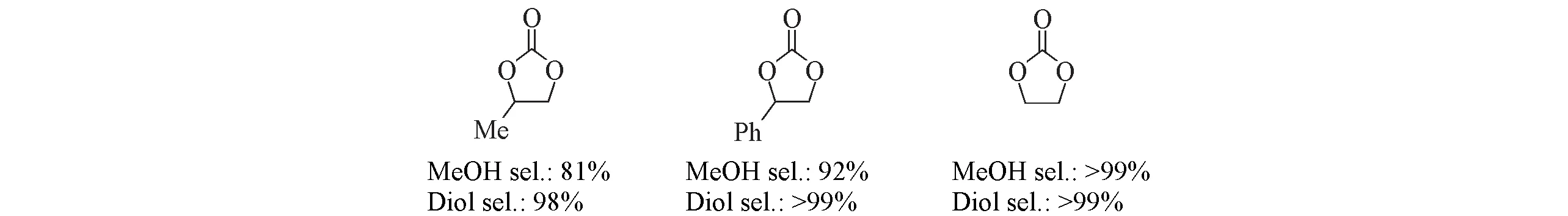
Scheme 4 Hydrogenation of various cyclic carbonates by manganese
均相催化剂在较低温度(120~140 ℃)下表现出较好的EC加氢催化活性,但均相体系存在分离困难的问题,且Ru基催化剂价格较贵,一定程度上限制了其大规模的工业应用.
1.2 多相催化剂
由于均相催化剂存在成本高、难于分离等问题,研究者对EC 加氢反应的多相催化体系开展了研究(Scheme 5),因金属Cu 裂解C—O 和C=O 的能力很强,所以该催化体系主要为Cu基催化剂[52,84].
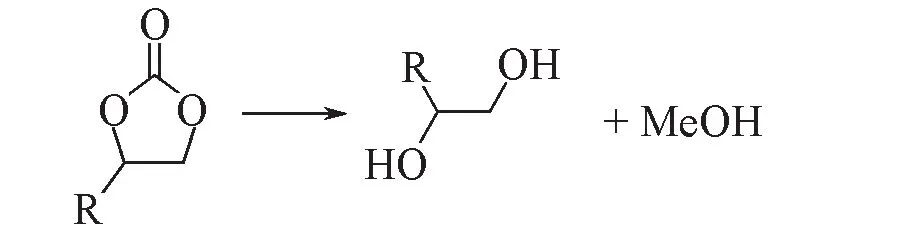
Scheme 5 Hydrogenation of cyclic carbonate
李亚栋课题组[77]通过水热法制备得到了CuCr2O4催化剂,并将其用于催化EC加氢体系,实现了EC加氢制备甲醇的多相化. 他们对其催化机理进行了探究,他们认为拥有四方尖晶石结构的部分CuCrO4,被还原成为具有立方晶型的金属Cu单质,促进了EC中C=O键的活化解离,从而得到了良好的催化性能. 这证明了Cu基催化剂对EC加氢有显著的催化作用. 但因为Cr元素有毒,因此发展绿色高效的无Cr催化剂十分重要.
陈静团队[78]采用浸渍法以及溶胶-凝胶法制备了多种Cu基催化剂,然后考察了其催化EC的加氢性能. 研究表明,催化剂中Cu颗粒尺寸(8~10 nm)适中、分布均匀分散,较高的比表面积、合适的酸碱性及适当的Cu0/Cu+比例,是该催化剂具有良好催化效果的原因;同时,随着酸性和碱性位点数量的增加,EC的转化率和甲醇的选择性几乎呈线性下降,但是催化活性与催化剂的酸性或碱性浓度之间没有明显的相关性,这表明过量的酸性和碱性位点的存在不利于EC高效产生甲醇. 此外,该团队提出了环碳酸酯加氢制备甲醇反应的机理(Scheme 6):首先,Cu0与Cu+协同催化环碳酸酯加氢开环,为该反应的决速步骤,吸附于催化剂表面的Lewis酸性位点的C=O受到Cu0上活泼的H进攻,得到中间体乙二醇单甲酯;乙二醇单甲酯继续加氢生成甲醇和乙二醇;同时乙二醇单甲酯可以在酸碱催化作用下,与生成的甲醇发生酯交换反应,生成甲酸甲酯和乙二醇,甲酸甲酯可以继续加氢生成甲醇;同时,生成的乙二醇单甲酯和甲酸甲酯可以在金属和酸碱催化作用下分解成乙醇、乙二醇、CO2和CO 等副产物[85,86],当催化剂表面酸碱性升高时,使该分解反应的速率迅速上升,导致甲醇选择性下降;除此之外,乙二醇可以在催化剂作用下缓慢氢解脱水生成乙醇.
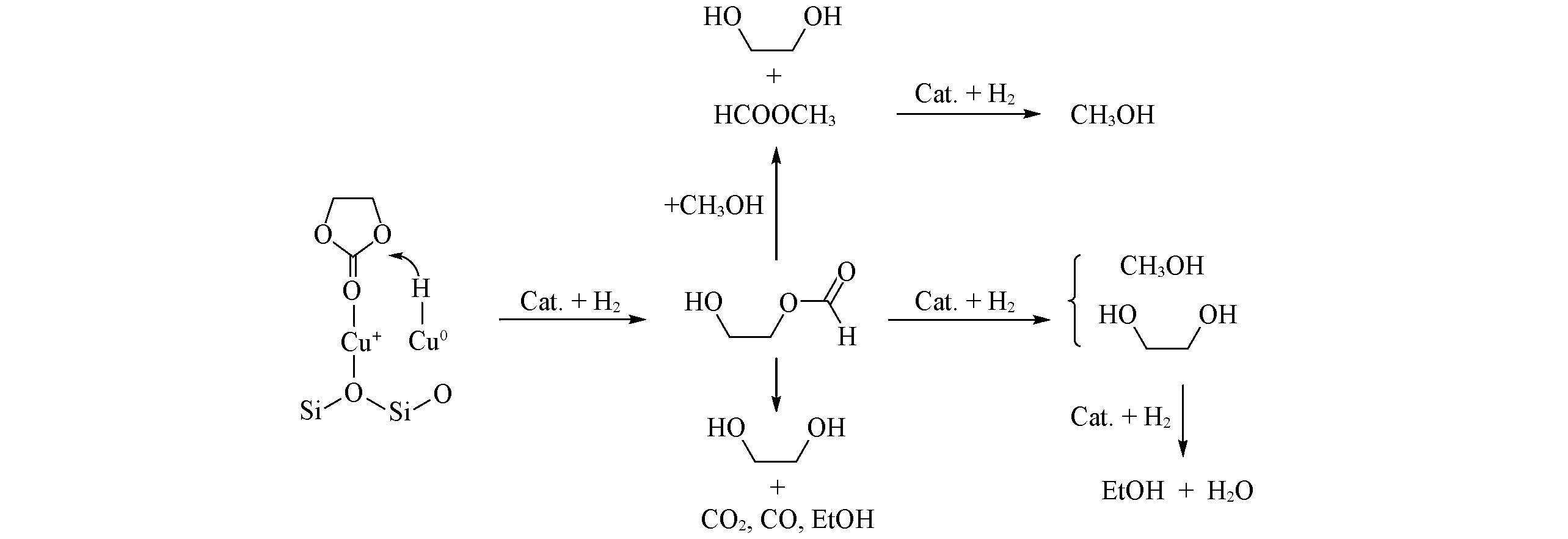
Scheme 6 Reaction mechanism of EC hydrogenation on Cu⁃SiO2 catalysts
戴维林团队[79]采用蒸氨法制备了不同Cu 负载量的Cu/HMS(Cu 质量分数20%,30%,40%,50%,60%)催化剂,然后对催化EC 加氢的催化性能进行了考察. 结果表明,50Cu/HMS(Cu 负载量50%)催化剂的催化效果较佳. 研究表明,催化剂中的Cu0和Cu+活性位点的协同作用会促进EC加氢反应,其中Cu0吸附和活化H2,Cu+作为甲氧基和酰基物种的稳定剂,同时Cu+位点可以作为亲电或路易斯酸性位点,通过氧中的孤电子对极化C=O键,从而提高酯基的反应性.
李会泉团队[80]采用一步水解沉淀法,以P123 为模板,制备了不同Cu 负载量的β-环糊精改性的xCu@SiO2-β-P(x=5,15,25,35,45)催化剂,并对催化性能进行了考察. 其中,使用P123作为模板剂是为了获得更大的表面积. 同时,他们提出了EC的加氢机理:EC首先被还原为甲酸-2-羟乙酯和1,3-二氧戊环-2-醇;随后,将甲酸-2-羟乙酯还原为乙二醇和甲醛;最后,甲醛中间体氢化形成甲醇.
岳海荣团队[81]采用沉积-沉淀法制备了不同Cu/Si 质量比的Cu/MCM-41 催化剂并用来催化EC 加氢. 结果表明,在Cu/Si 质量比为0.7时,催化活性最佳,并在固定床上研究了其催化加氢活性. 表征结果显示,Cu/Si 质量比会影响Cu0和Cu+的形成,Cu 颗粒会发生烧结而增大,并且Cu0/Cu+的比例发生了改变,会导致催化活性降低. 同时他们指出了两种价态Cu粒子的来源:Cu+源于铜氨络合物与载体表面的Si—O键相互作用,然后通过煅烧脱水形成硅酸铜;Cu0源于铜氨络合物在沉积-沉淀过程中除去NH3形成Cu(OH)2,煅烧后形成CuO,最后两者分别还原为Cu+和Cu0,其中Cu+在该还原温度下不会被还原为Cu0.
李晓红团队[82,87]用水热法制备了不同金属Mo担载量的Cu-MoOx催化剂,并将其应用于EC加氢反应. 实验结果表明,Cu-0.02MoOx/SiO2展示了最优的催化性能. 经过Mo掺杂后,Cu在催化剂载体上的分散度得到了提高,从而改善了其稳定性和对甲醇的选择性. EC 的转化率和MeOH 的选择性取决于Cu-mMoOx/SiO2催化剂中的Mo/Cu摩尔比. 根据表征结果,MoOx对Cu的改性不仅影响了Cu-mMoOx/SiO2的理化性质,也改变了Cu和Mo的电子特性. 这种改性通过Cu和MoOx之间的强相互作用,创造了具有Cu—O—Mo 键的界面位点,相应地改变了EC 的吸附和活化,并显着提高了MeOH 选择性. 特别是,Cu-MoOx界面处的Cu颗粒和MoOx之间的强相互作用,可以稳定Cu纳米颗粒并抵抗烧结,从而获得更好的催化剂稳定性.
2 环碳酸酯的醇解反应
环碳酸酯和醇在生成二取代碳酸酯的同时,可联产乙二醇(EG)或丙二醇(PG),因此,这是一条绿色经济的CO2间接转化路线,也是迄今为止商业化合成二取代碳酸酯最具前景的合成路线(Scheme 7).
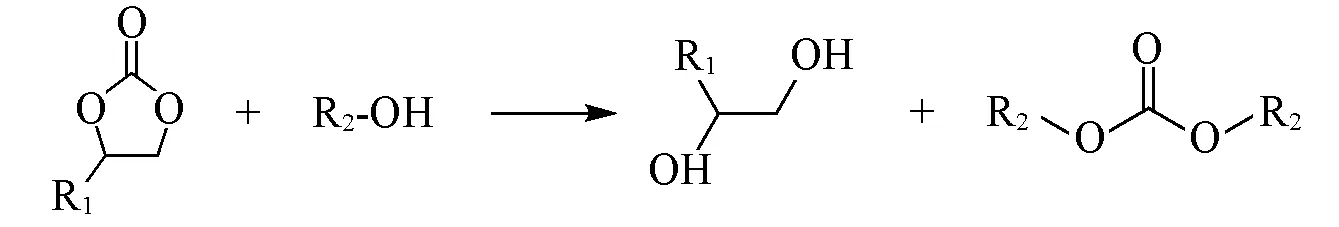
Scheme 7 Transesterification reaction formula of EC and alcohol
迄今为止,研究人员已开发了多种用于酯交换法合成二取代碳酸酯的催化剂,如离子液体、改性分子筛、单或混合金属氧化物、碱性阴离子交换树脂、蒙脱石、水滑石及其衍生物、片钠铝石、双金属氰化物、负载型催化体系和石墨氮化碳. 催化剂的活性和选择性如表2所示[88~108].

Table 2 Catalytic system of transesterification reaction between cyclic carbonate and alcohol
EC的醇解反应中最具发展前景的是,EC与甲醇进行交换合成碳酸二甲酯(DMC),同时联产乙二醇. 碳酸二甲酯是一种低毒、环保性能优异、用途广泛的化工原料,同时也是一种重要的有机合成中间体,分子结构中含有羰基、甲基和甲氧基等官能团,具有多种反应性能,在生产中具有使用安全、方便、污染少及容易运输等优点.
环碳酸酯的醇解反应属于典型的酸碱催化反应,即酸性催化剂和碱性催化剂都可以催化这个反应,但从目前已有的研究来看,碱性催化剂的催化活性相对较好. 其中,固体碱的主要功能是从醇分子中提取H+形成烷氧化合物. 因此,碱的强度决定了反应的速率,而碱性位的数量决定了反应的速率和产物的收率[109].
2.1 均相催化剂
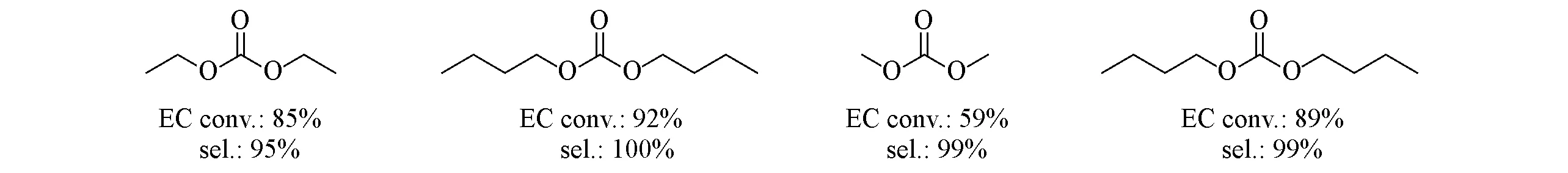
戴猷元团队[110]研究了碳酸钾催化的EC和甲醇的酯交换反应,发现超临界二氧化碳能降低副反应的发生,从而提高反应的选择性. 另一方面,二氧化碳的高压减缓了反应速率,降低了EC的转化率.在高压下EC的转化率和DMC的选择性可分别达到47.9%和98.1%.
Park团队[111]采用咪唑盐离子液体催化剂,研究了碳酸亚丙酯与甲醇的交换反应合成碳酸二甲酯.分别研究了不同烷基(C2,C4,C6,C8)和阴离子(Cl−,Br−,BF4,PF6)的1-烷基-3-甲基咪唑盐用于催化剂. 反应在140~180°C的高压釜中进行,二氧化碳压力为1.48~5.61 MPa. 烷基链长较短、亲核阴离子较多的咪唑盐型离子液体,在碳酸丙烯酯(PC)和甲醇合成DMC反应中表现出较高的反应活性. PC转化率随反应温度和CO2分压的升高而增加. 离子液体催化PC与甲醇的酯交换反应对PC浓度为准一级反应,其活化能为63.6 kJ/mol.
2.2 多相催化剂
Pochetti团队[112]用碳酸钠和磷酸钠催化EC与甲醇反应. 其中,磷酸钠在该反应中表现出良好的催化性能,使EC在非常温和的条件下,在很短的时间内(1 h)达到平衡产率(64%). 同时,以PC为底物时产率只有EC的1/3,并用动力学对其进行了解释:在甲醇过量的情况下,PC与EC的活化能很相近,但反应速率常数却是EC的1/5,这可归因于受到了空间位阻的影响.
研究人员认为离子液体是环碳酸酯和醇反应的最有效催化剂,但其与产物分离困难,为了解决这一问题,人们将离子液体进行固定化. 李永昕团队[107]将含有氨基丙基咪唑和OH-的碱性离子液体固定在氧化石墨烯上,合成了GO-[Ap-im]OH催化体系. 该材料的层状结构和优异的分散性确保了底物与活性位点的良好接触,作为EC和甲醇、乙醇、正丁醇和苯甲醇的酯交换催化剂,可以实现较好的催化活性和较高的稳定性(Scheme 8). 通过实验发现,催化体系中的NH2基团和OH−可能是主要的活性成分.
Scheme 8 Transesterification of different alcohols and esters catalyzed by GO⁃[Ap⁃im]OH
高国华团队[108]以N,N-亚甲基双丙烯酰胺(MBA)为交联剂,通过键联尿素的咪唑离子液体单体与丙烯酸钠的聚合,制备了脲功能化聚(PILs)离子液体,并用于EC和CH3OH的酯交换反应(Scheme 9).Poly(urea-IL)在CH3OH/EC混合溶剂中的溶胀能力,使得催化体系呈现出多孔的结构,而这种多孔结构有助于充分分散活性位点,提高反应的催化活性. 研究发现,键联尿素的咪唑离子液体单体可作为氢键供体活化EC. 虽然离子液体是环碳酸酯和醇的酯交换反应最有效的催化剂,且其可以通过载体固定化来解决产物分离困难这一问题,但这同时也为制备增加了难度、增加了生产成本,从而限制了它的工业化应用.
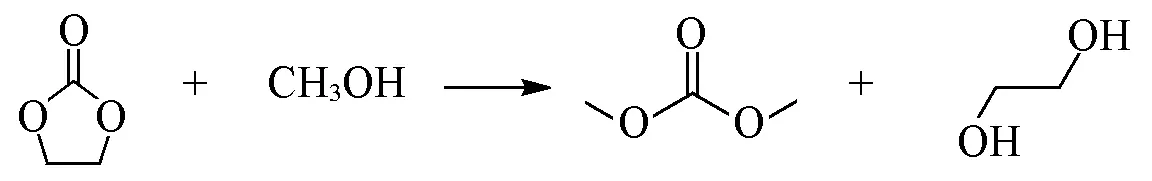
Scheme 9 Transesterification of methanol with EC
因为离子液体的高成本和繁琐的制备工艺,开发一种具有高催化活性和易于分离的新催化剂体系较为迫切. 研究者尝试开发了一系列的碱性金属氧化物以及混合金属氧化物催化体系. Ce是一种重要的稀土元素,已被广泛应用于汽车尾气处理的三效催化剂等催化工艺[113],其氧化物CeO2具有较强的碱性,研究人员认为这是一种极具前景的催化剂. 李永昕团队[97]以十六烷基三甲基溴化铵为模板,硝酸铈为前驱体,采用软模板法制备了介孔二氧化铈材料CeO2-meso-400,其表现出稳定的可回收性. 在此基础上,该团队还探索了由水热法合成的棒状、立方体状和多面体状等不同形貌的CeO2对酯交换反应的催化活性[105],其中,CeO2棒表现出最佳的催化活性(Scheme 10). 离子液体和其它二氧化铈基催化剂相比,非均相CeO2-meso催化剂在催化剂制备、回收利用和催化活性方面具有显著优势. 为了增强催化体系的活性,研究者用SrO,Y2O3,L2O3,MgO 等对CeO2进行修饰,合成SrO/CeO2[104],Y2O3/CeO2-La2O3[114],MgO-CeO2[91]等催化体系. 除此之外,研究人员还尝试了CaO-ZnO[115],ZnO2-Y2O3[95]等混合金属氧化物催化体系. 研究结果显示,催化剂的催化活性与催化剂表面碱度和碱强度分布有关,而与催化剂的比表面积无关.

Scheme 10 Transesterification of methanol with EC by CeO2
层状双金属氢氧化物具有较大的比表面积和较多的强碱性位点,可以作为环碳酸酯和醇的酯交换反应的催化剂. Nivangune团队[101]在Mg3-Fe LDH中掺杂适量的其它三价金属来调节LDHs的结构和化学性质. Ce的低电负性及适量的掺杂可以提高Mg3FexCe1-xLDH的碱性强度,从而使得催化活性有所提高. Gandara-Loe 团队[102]合成了以碳酸盐或硅酸盐为层间阴离子的XAl-LDHs(X=Mg,Zn,Ni)催化体系,其中,Ni基材料中碱位较多,其可能会在晶体上产生一些缺陷,从而提高催化活性. 研究人员还发现,当催化剂有酸性位点时,其产物选择性较低. 除此之外,该催化体系在重复使用时,需要在300 ℃下热处理6 h,这会造成更多的能源浪费. 除了上述的催化体系,研究者还合成了由稀土改性的Mg/Al水滑石(HT)煅烧制备的HT-10 La-C 三元氧化物催化体系[98],以及Cu-Zn-Al(CZA)水滑石催化体系[116]等,并发现这些水滑石或者类水滑石对环碳酸酯和醇酯交换反应也有催化活性. 这些具有层状结构的LDH催化体系大都是由共沉淀方法合成,制备方法简单,易于工业化生产.
虽然目前已经开发了多种环碳酸酯与醇酯交换反应的多相催化体系,但是还存在催化活性低、稳定性差、以及催化体系与产物分离困难等问题. 因此,研究者合成了Fe-Zn[117],Fe-Mn[106]等双金属氰化物催化体系. 这些催化体系具有较多的Lewis酸性位点,可活化醇并且协助环碳酸酯的开环. 与CaO等传统催化剂相比,该催化剂在反应过程中不易浸出且转化频率(Turnover frequency,TOF)值较高.
为了使催化体系具有更高的稳定性,除了上述的一些催化体系,负载型的催化体系也被用来催化环碳酸酯与醇酯的交换反应. 研究者将一些金属及金属氧化物负载到具有大的比表面积和孔结构的载体(如Au/CeO2[92],KF/Al2O3[93],MgO@meso-SiO2[96],Zr-Sn/GO[118]等)上. 相对于其它催化体系而言,负载型催化体系研究得较少,而且催化活性相对较低.
石墨相氮化碳(g-C3N4)因为含有大量的含氮基团,具有一定的碱性,而被应用于环碳酸酯与醇的酯交换反应. Xu等[99]通过一种简单、绿色的方法在g-C3N4中掺杂金属Zn,获得了催化活性较好、稳定性高的Zn-g-C3N4催化体系. 该团队认为,在EC与CH3OH的反应中,碱性位点的数量决定了催化体系的催化活性,而Zn-g-C3N4中的末端胺类和桥接胺类作为碱性基团提供了活性位点. 通过表征发现,Zn以离子态的形式存在,与g-C3N4存在相互作用. 随着Zn元素的引入,g-C3N4材料的边缘缺陷增加,其所含有的碱性位点的密度也有所增加,这也是Zn-g-C3N4具有较好催化活性的原因. 2017 年,李永昕团队[100]为了提高g-C3N4材料的碱性,在g-C3N4中引入了MgO,通过一种简单的混合煅烧法,成功地制备了MgO/g-C3N4(Scheme 11). 因为MgO的引入,使得三嗪基中间体分解,产生了更多的末端氨基,即有了更多的碱性位点,从而为反应提供了更多的活性位点. 该课题组合成的这两种掺杂金属的石墨相氮化碳催化体系,方法简单、绿色经济、催化活性和稳定性相对较好.

Scheme 11 Transesterification of methanol with EC by MgO/g⁃C3N4 catalyst
3 环碳酸酯的氨解反应
1,3-二取代脲已被广泛应用于农业生产、纤维染料、抗氧化剂、腐蚀剂以及药物中间体的制备等方面,这归因于其结构中具有生物活性的肽键(—CONH—). 传统合成1,3-二取代脲一般采用胺类与二氧化碳、碳酸二甲酯、一氧化碳和氧气、光气、异氰酸酯反应的方法. 但是因这些工艺需要有毒原料或较剧烈的反应条件而无法推广到工业生产中. 二氧化碳与胺的反应也可以产生二取代脲,但要获得较高的产率,则需要昂贵的脱水试剂(如碳二亚胺和有机亚磷酸酯等). 使用EC作为原料进行碱催化反应则可以避免以上问题(Scheme 12),从而利于绿色的工业化生产,催化剂的活性和选择性如表3所示[119~123].
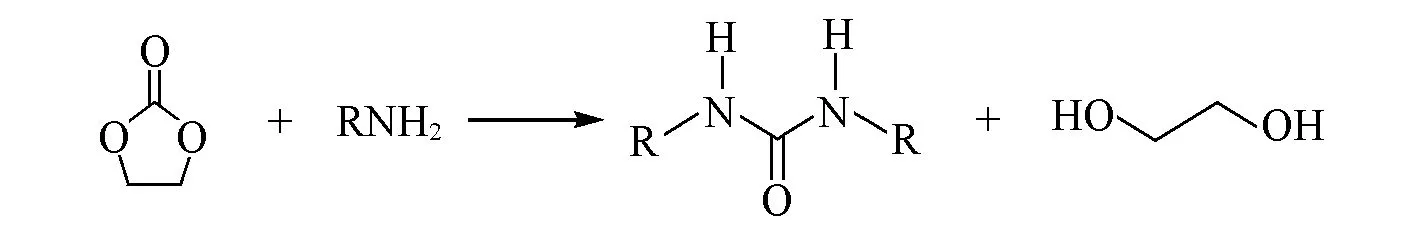
Scheme 12 Transesterification reaction formula of EC and amine
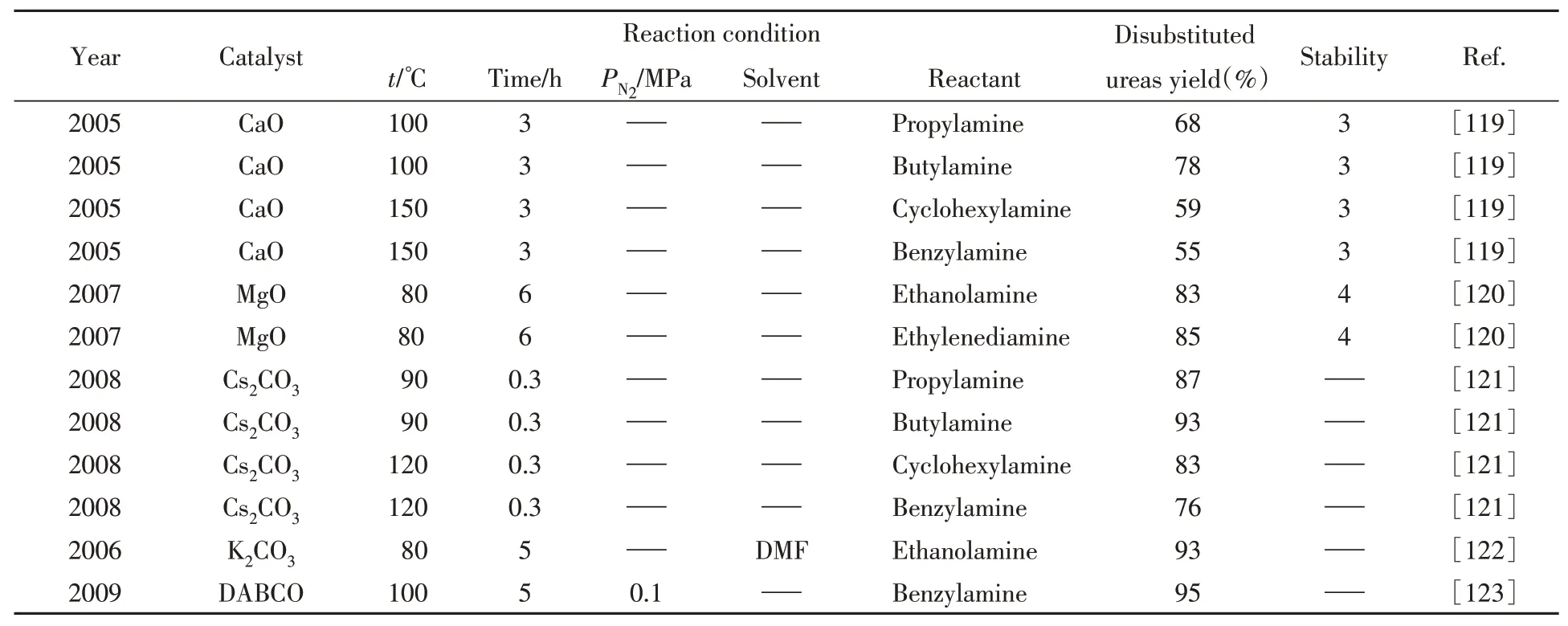
Table 3 Catalytic system of transesterification reaction between EC and amine
Arai团队[119]使用CaO作为催化剂,从EC制备1,3-二取代脲,并认为CaO的强碱性导致其具有高的催化活性(Scheme 13),同时提出了一种反应机理,确认了其中氨基甲酸酯和胺的催化反应是决速步骤. 该团队还以MgO作为催化剂[120],从EC和乙醇胺出发合成恶唑烷酮,获得了较高的产率.
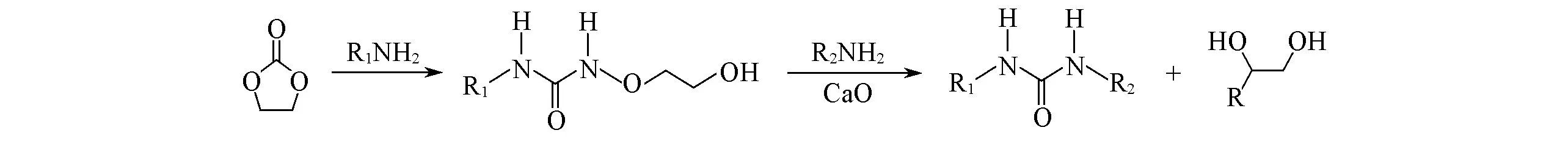
Scheme 13 Synthesis of unsymmetric disubstituted urea
Cs2CO3在该反应中的活性较好,受到了广泛的研究. Bhalchandra 团队[121]在温和条件下,使用Cs2CO3作为催化剂,催化EC和相应的胺高收率合成了1,3-二取代的对称/不对称脲.
恶唑烷酮在医药、化妆品、农药等有机合成中有着广泛的应用,它们可用作有机合成中的保护基团、不对称合成中的手性助剂(Evans的手性助剂)和生物活性试剂. 夏春谷团队[122]以K2CO3为催化剂,从2-氨基乙醇与碳酸丙烯酯制备2-恶唑烷酮. 该方法适用于由不同的β-氨基醇和五元环碳酸酯合成相应的2-唑烷酮(Scheme 14),几乎所有使用的β-氨基醇和五元环碳酸酯都表现出很好的产率.
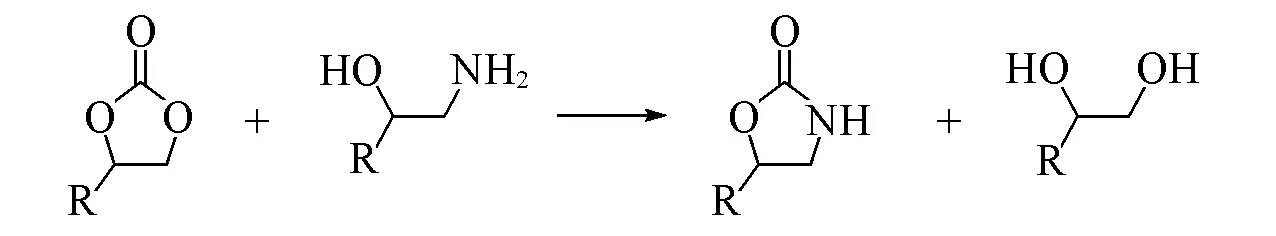
Scheme 14 Synthesis of 2⁃oxazolidone
邓国军团队[123]首次报道了在三乙烯二胺(DABCO)存在下,由EC和芳基胺合成恶唑烷酮,同时研究了苯环上取代基对该反应的影响(Scheme 15),结果表明,对位或间位含吸电子基团或不含取代基的芳基胺产率较高,若是供电子基团则产率略微下降,若邻位上有取代基则产率大幅下降,推测这是由于邻位取代基的空间位阻造成的.

Scheme 15 Synthesis of 3⁃phenyl⁃2⁃oxazolidinone
4 总结与展望
环碳酸酯作为CO2间接转化制备其它化学品的反应中间体,不仅可以避免CO2直接利用中由于化学惰性所带来的反应条件苛刻等不利因素,而且能够有效实现CO2产业链的拓展,从而获得高附加值精细化学品. 本文分别从环碳酸酯的加氢反应、醇解反应和胺解反应等3方面介绍了其研究进展.
到目前为止,在环碳酸酯的加氢反应中,Cu基催化剂展现出优异的催化性能,但在长期的稳定性考察过程中发现,催化剂存在易失活等问题,且该类催化剂的选择性仍待进一步提升,应从调节活性金属的电子密度、增强金属-载体相互作用等方面进行考虑,以及对催化剂的设计和制备方法进行调整,以期提升催化剂的稳定性和对产物的选择性. 目前,使用C,B,Zn,Au等元素对Cu基催化剂进行掺杂,可以有效提升Cu基催化剂的活性和增强其稳定性[124~129].
在环碳酸酯和醇交换生成二取代碳酸酯反应体系中,固定化的离子液体解决了产物分离困难这一问题,但这同时也为制备增加了难度,增加了生产成本,从而限制了其工业化应用. 虽然双金属氰化物与石墨相氮化碳等催化体系中的酸碱位点可以提供高活性的位点,然而该反应的另一产物乙二醇在酸碱位点的作用下会分解生成水和乙醇,使其无法被回收利用,降低了反应的经济性. 因此,需要开发新的催化体系,以期同时获得二取代碳酸酯和二醇,从而实现反应的高值化.
在环碳酸酯与胺交换生成1,3-二取代脲或恶唑烷酮反应体系中,碳酸铯的催化活性最高,而氧化钙可以回收利用,但反应的活性不如前者. 对于环己胺和仲胺,在同等反应条件下,产率不及伯胺高,这可能是位阻原因所致. 同时,该反应中的催化机理构效关系尚未得到深入研究,还需要通过理论与原位实验相结合的手段进一步加以印证.
CO2间接转化为环碳酸酯,再转化为甲醇等小分子化合物或其它精细化学品,既具有经济意义,又具有环保意义. 但是目前对于这类反应的研究大多数还处于实验室阶段,反应机理不够明确,产物选择性和底物转化率均不太理想,且成本过高,不能进行商业化推广. 制备高活性的催化体系,实现CO2转化为环碳酸酯,继而间接转化为高附加值的化工产品,对解决环境问题、能源问题和促进化工的产业升级均具有重大的意义.
猜你喜欢
科学大众(2023年17期)2023-10-26 07:38:56
小天使·二年级语数英综合(2021年5期)2021-07-11 10:58:35
中国油脂(2020年5期)2020-05-16 11:23:52
中学化学(2017年2期)2017-04-01 08:51:54
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
无机化学学报(2014年7期)2014-02-28 17:32:10
无机化学学报(2014年4期)2014-02-28 17:31:23
应用技术学报(2014年1期)2014-02-28 14:52:11
食品工业科技(2014年12期)2014-02-28 08:09:59
