
多孔骨架固定分子催化剂催化CO2加氢制备甲酸研究进展
2022-08-06 04:37王万辉
高等学校化学学报 2022年7期
丁 杨,王万辉,包 明
(1.大连理工大学精细化工国家重点实验室,大连 116023;2.大连理工大学化工学院,盘锦 124221)
自工业革命以来,化石燃料作为社会经济发展的主要能源被广泛使用,释放了大量温室气体二氧化碳(CO2),导致了全球变暖和生态破坏等严重的环境问题[1]. 二氧化碳捕集和利用不仅可以消减二氧化碳,还可以提供有价值的产品,吸引了越来越多的关注. CO2是一种无毒、廉价且丰富的C1资源,可以转化为多种高附加值的工业产品,例如甲酸、甲醇和碳酸酯等[2~4]. 其中,甲酸(FA)是最重要的产品之一. 甲酸是基本的有机化工原料,在医药、农药、皮革、食品、橡胶、化学合成和新能源材料等领域有广泛的应用[5~7]. 在新能源材料方面,甲酸可以用作有机液体储氢材料,它的体积氢容量高达53 g/L,超过了多种氢化物和压缩氢气的氢容量(20~40 g/L). 另外,甲酸作为能源材料可以应用于直接甲酸燃料电池(DFAC)提供电力[8]. 利用CO2加氢制备FA并应用于储氢和燃料电池领域有助于开发绿色氢能源和消减二氧化碳,助力我国“双碳”目标达成[9].
二氧化碳具有极高的热力学稳定性和动力学惰性. CO2加氢合成甲酸在热力学上是不利的,气态反应的标准吉布斯自由能为32.9 kJ/mol[式(1)]. 添加碱吸收反应产生的质子能够促进反应进行[式(2)][10]. 由于CO2自身的固有惰性以及CO2加氢生成甲酸的热力学不利因素,开发经济高效的CO2转化催化剂,实现CO2向甲酸的有效转化成为当前该领域面临的重大挑战.
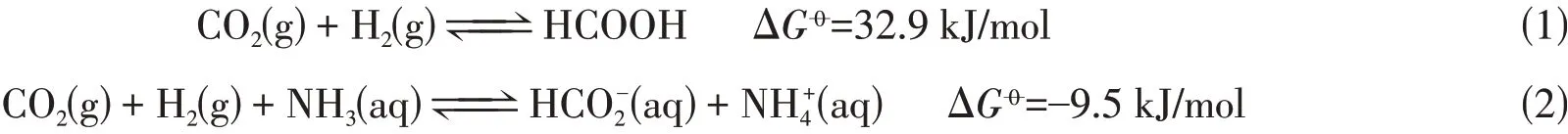
三苯基膦钌配合物均相催化CO2加氢生成甲酸的研究首次于1976年报道[11],而后均相催化剂的研究日益兴盛. 尤其是近30年来均相催化剂的研究取得了重要进展[12,13],其中以半夹心型配合物和三齿钳合物的催化活性最佳[14]. Himeda 课题组[15,16]开发的一系列含功能基团的氮杂环半夹心型金属配合物(1~3,图1)在水体系中于温和的条件下就可以催化CO2加氢制备甲酸的反应,其中催化剂2的转化数(TON)达到79000. 配体上邻位羟基侧坠可以辅助H2分子异裂,促进Ir-H中间体的形成,从而大大提升催化性能. 李灿课题组[17]开发的N,N双齿半夹心催化剂(4,图1)在无碱条件下展示出优异的活性,转化频率(TOF)高达13109 h−1. Nozaki等[18]和Pidko等[19]报道的铱、钌钳合物(5~8,图1)在有机体系中展示出极高的活性,催化剂5的TON高达3500000,催化剂8的TOF高达110000 h−1. 这些强供电子配体或功能化配体可以通过电子效应或侧坠效应等进行调控,极大地提升了催化剂的活性.
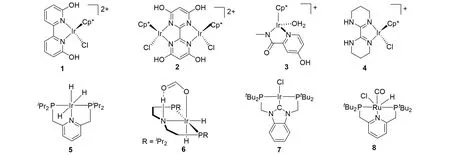
Fig.1 Highly active homogeneous catalysts for the hydrogenation of CO2 to formic acid[15~19]
均相催化剂尽管具有极高的活性,但是存在难以分离回收的问题. 为了结合均相催化剂高活性和高选择性的优势以及多相催化剂便于分离和循环使用的优点,均相金属配合物的多相化研究逐渐发展起来. 将Ru/Ir配合物固载到传统的二氧化硅微球或聚合物载体上制备的催化剂具有良好的回收性能,验证了这一策略的可行性[20~22],但是在催化活性和稳定性上依旧有待提高. 近年来,具有比表面积大、孔径明确、方便使用和合成路线多样化等诸多优点的新型骨架材料,如共价有机框架(COFs)、多孔有机聚合物(POPs)和金属有机框架(MOFs)等在气体吸收分离、催化等领域引起了广泛关注[23~26]. 这类骨架材料多以有机物作为连接体,便于修饰和调节,方便引入配位点,为新型催化材料的开发提供了新的契机. 近年来,以此类骨架材料与均相催化剂相结合构建新型的多相催化剂用于CO2加氢还原制备甲酸取得了重要进展. 本文对不同骨架负载的催化剂的研究进展分别展开介绍,并对其在CO2加氢还原制备甲酸领域的应用前景进行了展望.
1 COF负载型催化剂
共价有机框架是近年新兴的一种结晶有机多孔材料,具有二维或三维多孔晶体结构,材料的组成、拓扑和孔隙率易于调控. 由于COF的结构单元之间通过牢固的共价键相连,材料具有超高的化学和热稳定性[27]. 自2005年,Yaghi课题组[28]首次报道硼酸酐连接的COF材料以来,经过约20年的不断深入研究,COF材料已经逐渐发展为一类重要的有机功能材料[29]. 根据连接方式的不同,可以把COF分为含硼型、亚胺型、三嗪型、酰胺型、酰亚胺型、苯腙型、酰肼型、碳-碳双键共轭型和芳香醚型等.由于具有明确的晶体结构、较大的表面积、有序的孔道结构、良好的化学稳定性以及易于功能化修饰等突出特点,其在CO2加氢领域表现出巨大的应用潜力.
2015 年,Yoon 课题组[30]合成了含有三嗪和联吡啶单元的共价三嗪框架(Bpy-CTF),并将其与[IrCp*Cl2]2前体反应,制备了二维Ir@bpy-CTF 骨架催化剂. 联吡啶结构与Ir(III)发生强螯合,实现了高含量的Ir 负载(质量分数约4.7%). X 射线光电子能谱(XPS)结果证实Ir/Cl 原子比接近1∶2.4,并且表明Ir与给电子配体结合. 在120 ℃,8 MPa[V(CO2)∶V(H2)=1∶1]的总压下,在1 mol/L的三乙胺(Et3N)水溶液中催化CO2加氢制备甲酸盐,2 h 后TON 达到5000,初始TOF 达到5300 h−1. 但其回收利用性能较差,在第5 次循环后,TON 从5000 降到3000,有较大损失. 浸出实验表明有5.5%的金属浸入溶液中. 该课题组[31]进一步研究了二维Rh@bpy-CTF催化剂(图2). 在同样条件下,二维Rh@bpy-CTF催化剂的TON为1410,TOF为960 h−1,比Ir催化剂的活性大幅降低.
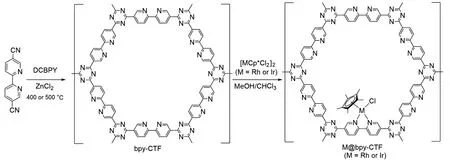
Fig.2 A general synthetic scheme for the preparation of bpy⁃CTF⁃based Rh and Ir complexes[31]
2016年,Yoon课题组[32]开发了二维层状有序庚嗪基骨架(HBF),在固定Ir(III)后得到非常有效的COF 催化剂HBF-Ir,用于将CO2加氢生成甲酸盐. 在8 MPa[V(CO2)∶V(H2)=1∶1],120 ℃,1 mol/L 的Et3N 水溶液中反应10 h,TON 可达6400,TOF 可达640 h−1. 作者将HBF-Ir 催化剂与含有庚嗪单元的“C3N4”进行了对比(图3),发现含庚嗪的HBF-Ir 催化剂具有更高的活性和更好的可回收性. 一方面,HBF-Ir中更多的介孔(平均孔径为19.6 nm)有利于底物更好地扩散到活性位点;另一方面,HBF-Ir显示出远优于均相IrCl3的活性,这是因为骨架内的Ir(III)离子具有更高的电子密度,表明庚嗪和Cp*配体向Ir阳离子提供了电子. HBF-Ir催化剂可循环使用3次而不发生明显的活性损失.
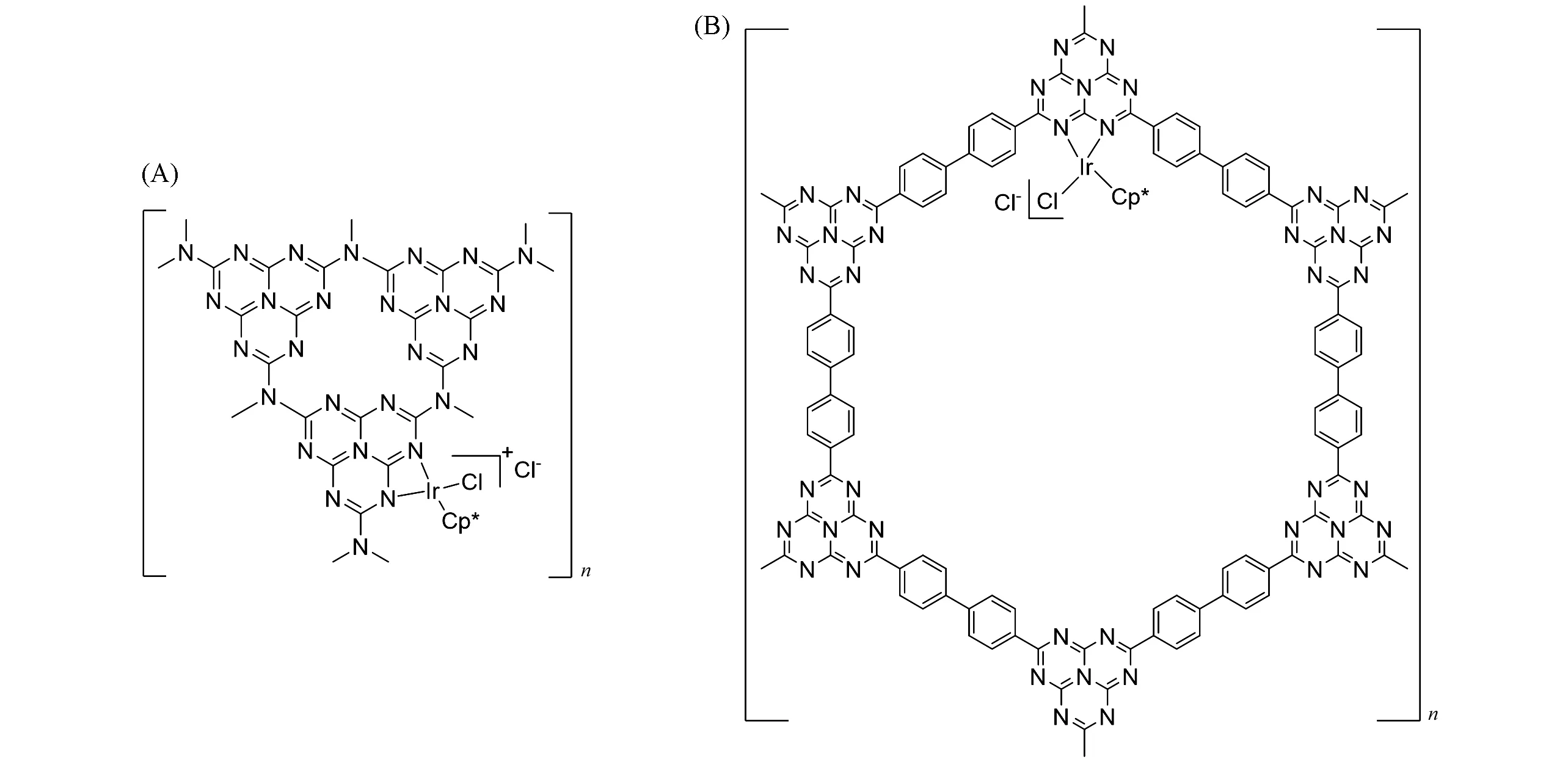
Fig.3 Structures of Ir⁃C3N4(A)and heptazine⁃based COF containing Ir(III)complexes(B)[32]
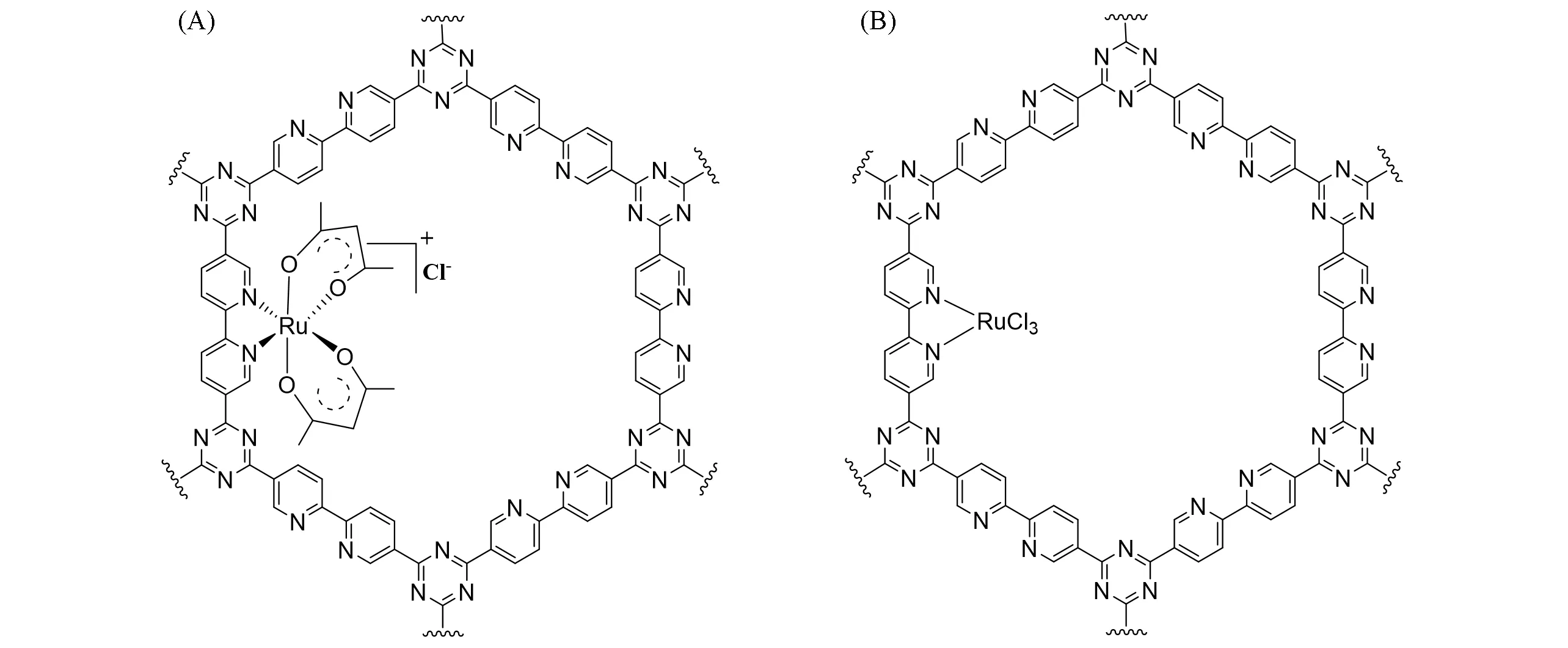
Fig.4 Structures of[bpy⁃CTF⁃Ru(acac)2]Cl[33](A)and[bpy⁃CTF⁃RuCl3][34](B)
为了解决Cp*M型金属中心的浸出问题,2018年,Yoon课题组[33]使用乙酰丙酮(acac)代替Cp*,合成了多孔[bpy-CTF-Ru(acac)2]Cl催化剂[图4(A)]. 他们认为Ir金属中心催化H2异裂时,联吡啶氮会发生质子化进而发生解配,因此利用乙酰丙酮捕获H2异裂产生的质子,从而避免金属中心与吡啶配体的解配,进而减少金属浸出. 该催化剂在1 mol/L的Et3N水溶液中,于8 MPa[V(CO2)∶V(H2)=1∶1],120 ℃条件下展示出极高的初始TOF(22700 h−1);而在3 mol/L 的Et3N 水溶液中,于8 MPa[V(CO2)∶V(H2)=1∶1],120 ℃条件下反应5 h 的TON 高达21200. 该催化剂展示出优异的回收性能,重复利用4 次后,催化剂的活性没有降低. 含氧配体的引入不仅提升了催化剂的活性,同时也大大改善了催化剂的回收利用性能. 2019 年,该课题组[34]将RuCl3负载在含有三嗪和联吡啶单元的共价三嗪框架上,得到[bpy-CTF-RuCl3]催化剂[图4(B)]用于CO2加氢生成甲酸. 在120 ℃,8 MPa[V(CO2)∶V(H2)=1∶1],3 mol/L 的Et3N水溶液中反应2.5 h,TON可达20000,TOF可达38800 h−1. 该类催化剂具有出色的回收性能,可循环使用5次而不发生明显的活性损失. 基于DFT计算的机理研究表明,高压下CO2在Et3N水溶液中形成碳酸氢盐,碳酸氢根取代acac配体并形成[Ru(acac)(HCO3)(bpy)]中间体. 该中间体随后与H2反应形成Ru-H活性中间体. 该步骤为反应的决速步骤. 随后,CO2插入到Ru—H键之间,形成了甲酸根配位的中间体. 甲酸根解离后完成整个催化循环.
2016 年,Gascon 课题组[35]制备了一种可充分回收的CTF 基球体Ir@CTF 催化剂,并用于在温和条件下催化CO2加氢直接合成甲酸. 他们以聚酰亚胺Matrimid 5218 作为黏合剂,通过相转化法将介孔CTF嵌入到Matrimid 5218中,并负载Cp*IrIII制备出稳定的CTF基球体Ir@CTF催化剂,其结构如图5(A)所示. Ir含量为2.4%的球体Ir@CTF催化剂在2 MPa(H2/CO2)和90 ℃条件下,在1 mol/L KHCO3水溶液中反应2 h,催化CO2加氢生成甲酸盐的TON可达358. 球体Ir@CTF催化剂在循环回收后质量损失小于5%,且在第4次循环后仍保持高稳定性和活性. 2019年,Urakawa 课题组[36]以2,6-二氰基吡啶为原料通过离子热聚合制备了CTF骨架材料,然后负载[IrCp*Cl2]2合成了一种多相Ir-CTF催化剂[图5(B)].在100 ℃、30 MPa 条件下,在三乙胺存在下连续反应48 h 合成甲酸盐,总TOF 为1.61. 对新鲜催化剂和使用后的催化剂进行STEM和EDX表征,证实了反应前后Ir均匀分散. XRD测试结果表明,反应后的催化剂在2θ=12°附近的衍射峰消失,表明在反应过程中配合物有浸出. ICP-OES测试结果表明,反应后催化剂的Ir含量从最初的16.3%降低到7.6%,证实了反应过程中Ir的损失.
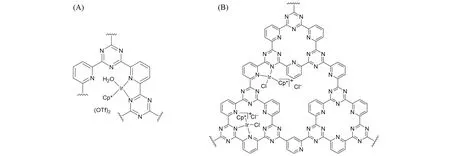
Fig.5 Structures of CTF⁃based sphere Ir catalyst[35](A)and Ir⁃CTF[36](B)
2 POP负载型催化剂
近年来,多孔有机聚合物发展迅速,主要类型包括:多孔芳香骨架材料(PAFs)、超交联聚合物(HCPs)、固有微孔聚合物(PIMs)和共轭微孔聚合物(CMPs)等. 与COFs不同的是,POPs通常是由不可逆反应形成的无定形材料,其中的孔结构分布相对较乱,没有规律性. POPs也有极高的孔隙率、比表面积和稳定的共价键,尤其是富含可与活性金属有效结合的功能位点,这些对催化CO2分子转化都非常重要[37].
2015 年,刘志敏课题组[38]制备了含有Trögers 碱(TB)作为内置基序的微孔有机聚合物(MOP),随后固载Ru(III)配合物形成Ru-TB-MOP 框架(图6). TB-MOP 具有极大的BET 比表面积(802 m2/g)和孔容(0.5 cm3/g),有利于Ru 的固载. TB-MOP(27.9 kJ/mol)和TB-MOP-Ru(28.5 kJ/mol)具有较高的CO2等量吸附热值,表明TB 功能化聚合物的亲CO2特性. 然而,该值仍低于化学吸附过程的能量(>40 kJ/mol),表明可极化CO2分子可能与TB上的N原子发生强烈的相互作用. 其在5 mL Et3N和1 mmol PPh3存在下,在12 MPa H2/CO2总压和40 ℃条件下反应24 h的TON为2254. 催化剂可循环使用3 次而不发生明显的活性降低. 该研究揭示了TB的双重功能:既可以与Ru 形成稳定的配合物,又能够吸附CO2以确保反应中心的高CO2底物浓度.

Fig.6 Synthetic route of the TB⁃functionalized MOP(TB⁃MOP)[38]
2018 年,Palkovits 课题组[39]报道了固载在含膦聚合物(pDPPE)上的Ru配合物催化CO2加氢制备甲酸盐的反应. 金属载量为0.5%的Ru@pDPPE催化剂在10 MPa[V(H2)∶V(CO2)=1∶1]总压和120 ℃下,在2.59 mol/L K2CO3水溶液中反应4 h,TON可达13170. 回收实验显示第一轮反应后催化活性明显降低. 结构表征显示Ru浸出率比较低,而且pDPPE的结构没有明显变化. 研究发现Ru活性中心的对甲基异丙苯配体发生了解离,这可能是导致催化剂活性降低的原因. 同时还发现Ru催化甲酸分解会产生CO,它有可能使催化剂活性降低. 基于实验结果提出了可能的反应机理(图7). 首先,催化剂A 加氢后脱除氯配体生成氢化物B;随后,对甲基异丙苯配体被CO 取代,生成中间体C,CO2插入RuII—H键中,形成甲酸根配合物D;接下来,经历氢分子的氧化加成和甲酸的还原消除使活性中间体C再生,同时,生成的甲酸被碱夺去质子生成甲酸盐.
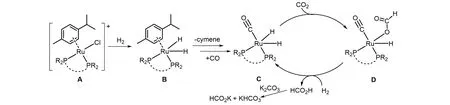
Fig.7 Proposed mechanism for the catalytic hydrogenation of CO2 to formate[39]
2019 年,Yoon 课题组[40]通过Friedel-Crafts 聚合法合成了含邻菲罗啉单元的多孔有机聚合物(phen-POP),合成途径如图8所示. 邻菲罗啉对付克烷基化高度不活泼,因此使用苯基取代的邻菲罗啉衍生物作为单体来构建phen-POP 骨架. 骨架中含有大量N,N 位点,可以与金属络合并将其固定在载体骨架上. 骨架负载氯化铱(III)后得到多相IrCl3-phen-POP 催化剂,并用于将CO2加氢成甲酸盐.IrCl3-phen-POP 在8 MPa、140 ℃、1 mol/L Et3N 水溶液条件下反应2 h,TON 可达14330,初始TOF 可达40000 h−1,且循环使用3次后活性无明显降低.
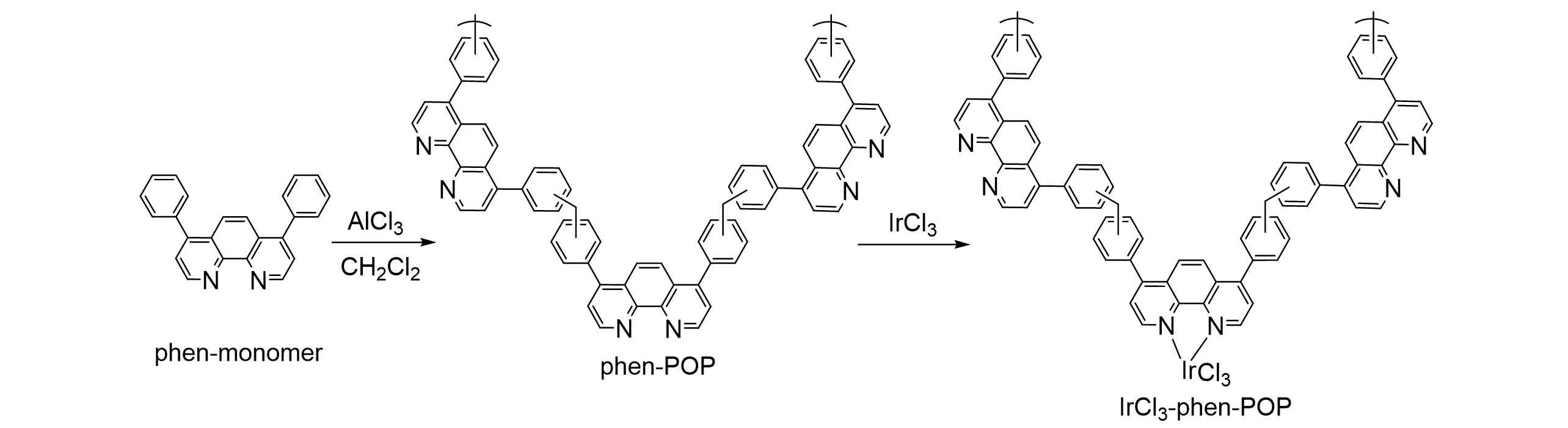
Fig.8 Schematic representation for the novel route of synthesis of IrCl3⁃phen⁃POP catalyst[40]
2019年,黄延强课题组[41]制备了含有吡啶单元的酰胺型多孔有机聚合物,并以其为载体负载Ir金属得到了准均相的单原子催化剂Ir/AP-POP,合成过程如图9 所示. 由于载体表面富含氮碱性位点,Ir/AP-POP 表现出与CO2的强相互作用. 在140 ℃、8 MPa 条件下,在1 mol/L Et3N 水溶液中反应24 h,TON 高达25135. 该催化剂的催化性能远高于以活性碳和氮化碳为载体的Ir 催化剂. 研究表明,Ir/AP-POP 骨架中吡啶单元的供电子特性对于提高Ir的活性至关重要. 2020年,邵先钊课题组[42]制备了吡啶和酰胺基多孔有机聚合物负载的Pd 催化剂Pd/AP-POP,在80℃、8 MPa 条件下,在Et3N 水溶液中反应12 h,TON为1279. 其催化性能明显高于均相的PdCl2配合物[43]. 研究证明了吡啶N位的双重作用:既作为碱性位点吸附CO2,加快反应速率,又能作为供电子基团增加金属的电子密度,提升催化活性.
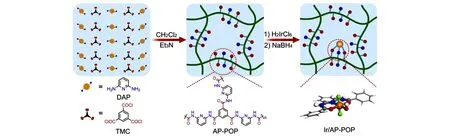
Fig.9 A schematic illustration of the synthesis of Ir/AP⁃POP single⁃atom catalyst[41]
2020 年,Yoon 课题组[44]制备了三聚氰胺网络聚合物(MPN)负载的RuCl3催化剂,并用于CO2加氢反应. 如图10所示,通过对苯二甲醛和三聚氰胺缩合制备MPN骨架,然后负载金属Ru,经真空干燥后得到Ru基催化剂. 在无碱、120 ℃及8 MPa 总压下反应2 h,TON 可达107,TOF 可达54 h−1. 在1 mol/L Et3N 水溶液中,于120 ℃及8 MPa 总压下反应15 min,TON 可达1242,TOF 可达4964 h−1. Ru-MPN 在0.63%的Ru含量、120 ℃及8 MPa总压下,于1 mol/L Et3N水溶液中反应2 h,得到最佳的TON 4492. 在相同反应条件下,当Ru 含量为0.32%和1.08%时,其TON 值分别为1895 和2932. 从反应结果推断,TON随着钌含量的增加而增加是由于聚合物上具有更多的活性金属中心位点,从而提高了CO2的转化率;然而,随着钌含量的进一步增加,碱性中心的饱和导致CO2吸附位点减少,从而使TON降低.

Fig.10 Synthesis of melamine polymer network and ruthenium impregnated catalyst[44]
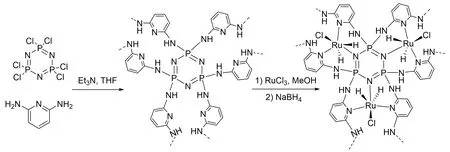
Fig.11 Schematic representation for the preparation of the p⁃dop⁃POM and Ru/p⁃dop⁃POM[45]
2020 年,韩布兴课题组[45]由廉价易得的三聚氯化磷腈和2,6-二氨基吡啶制备了多孔聚合物p-dop-POM(图11),并与RuCl3反应合成了多相催化剂Ru/p-dop-POM. 在8 MPa 总压和120 ℃条件下,在2.57 mol/L的Et3N水溶液中反应24 h,TON高达25400. 该催化剂不仅性能优异,而且循环使用3次无明显活性损失. DFT 计算表明,由磷腈N 和吡啶N 配位提升了Ru 中心电子云密度,可以促进H2解离,从而提高催化性能. 富电子Ru中心和富氮聚合物之间的协同效应对高催化性能至关重要.
2021年,Yamashita课题组[46]将含N、P的多孔有机聚合物包封在介孔空心碳球(MHCS)中,并负载RuCl3得到Ru3+POPs@MHCS 三元混合多相催化剂,用于催化CO2加氢生成甲酸盐. 在4 MPa 总压和120 ℃下反应24 h,TON超过1200,可循环使用3次而无明显的活性损失. MHCS和POP表现出较高的CO2吸附能力. 优异的催化性能归因于MHCS的高表面积和大孔体积,有利于Ru3+-POP的分散和稳定.
3 MOF负载型催化剂
金属有机框架是通过拼接多核金属簇和有机连接体作为二级结构单元而形成的具有周期性网络结构的晶体配位聚合物[47]. 由于其合成简单、具有高孔隙率和结晶度、结构可调、易于修饰等特点,MOFs作为CO2吸收和转化的多相催化剂具有广泛的前景[48]. 值得注意的是,MOFs中的不饱和金属位点对CO2还原反应具有重要的作用. 近年来,MOFs在CO2化学还原催化方面的应用显著增多[49].
2013 年,Limtrakul 课题组[50]报道了Cu 官能化MOF 催化CO2加氢制甲酸的理论计算研究结果. 他们选用Zn 基MOF-5 作为模板,在对苯二甲酸连接体上通过引入两个邻位酚羟基与Cu 配位构建Cu-MOF-5 催化剂(图12). DFT 计算研究反应机理显示有两种可能的反应途径,即协同机理和分步机理. 协同机理中CO2氢化成甲酸一步完成,但活化能高达67.2 kcal/mol(1 kcal=4.19 kJ). 分步机理中,CO2和H2在Cu-O 活性位点上吸附后,首先氢气异裂,负氢进攻CO2的碳原子形成甲酸根配位的中间体,然后甲酸根进一步从酚羟基上吸收质子生成甲酸. 第一步和第二步的活化能分别为24.2和18.3 kcal/mol. 该途径的活化能较小,因此分步机理比协同机理更有优势. 该研究还表明,在苯环上引入氨基供电子基团可以提升Cu 金属中心的电子云密度,促进Cu 金属和吸附分子CO2/H2之间的电子转移,从而降低反应能垒.
2015 年,Johnson 课题组[51]利用DFT 计算方法设计催化剂,将受抑路易斯酸碱对(FLP)结合到UiO-66型MOF中,作为CO2加氢还原制备甲酸的催化剂. 选用二氟硼基吡唑单元(P-BF2)作为路易斯酸碱对活性催化位点,将P-BF2与UiO-66的对苯二甲酸连接体接枝构建了P-BF2功能化的MOF催化剂,结构如图13所示. P-BF2路易斯酸碱对可以促进H2的异裂,从而加速CO2还原. 理论计算显示该催化剂的能垒比一般多相催化剂低,与均相催化剂接近. 该研究为高效CO2还原催化剂的设计提供了一种新策略.

Fig.12 CO2 hydrogenation to formic acid with Cu⁃MOF⁃5 catalyst[50]

Fig.13 Schematic of a UiO⁃66 primitive cell functionalized with P⁃BF2[51]
2017年,程俊课题组[52]将Ir(III)负载到UiO型MOF中获得了功能化的MOF催化剂mbpy-IrCl3-UiO和mbpyOH-IrCl3-UiO,其合成过程如图14 所示. 将1 mol/L 的NaHCO3水溶液置于下部蒸馏瓶中,催化剂置于索氏提取器中,在常压H2/CO2气氛、加热回流条件下催化反应. 当冷凝下来的热水滴渗入MOF 催化剂时,CO2、H2、H2O 和催化剂的接触面积最大化,从而构建了良好的催化反应体系.mbpyOH-IrCl3-UiO 催化剂在85 ℃和0.1 MPa H2/CO2(体积比1∶1)条件下反应15 h 后,TON 达到6149,TOF 达到410 h−1;而在相同条件下,mbpy-IrCl3-UiO 催化剂的TON 只有417,TOF 为28 h−1. 这与同类型均相催化剂的反应结果类似,证明含有邻位羟基的催化剂活性更高. 理论计算证实邻位羟基能够在氢气异裂过程中与Ir金属中心协同作用,促进质子传递.
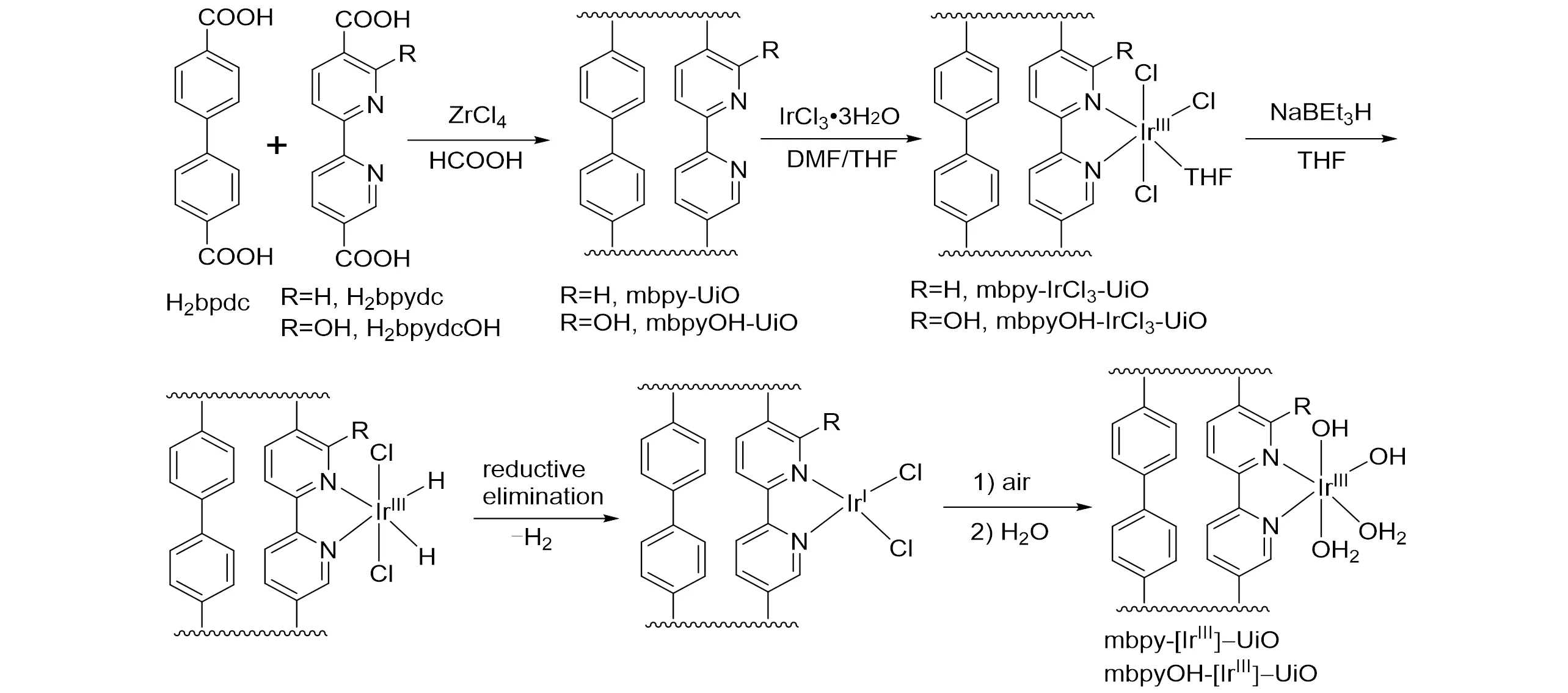
Fig.14 Preparation of mbpyOH⁃[IrIII]⁃UiO and mbpy⁃[IrIII]⁃UiO[52]
2019 年,赵玉军课题组[53]利用合成后修饰的方法将水杨醛和2-二苯基膦基苯甲醛(DPPBde)与MIL-101(Cr)-NH2的末端氨基反应后固载Ru(III),成功合成了高效的RuCl3@MIL-101(Cr)-Sal 和RuCl3@MIL-101(Cr)-DPPB 催化剂. RuCl3@MIL-101(Cr)-DPPB 中强供电子的N,P-配体和Ru(III)离子具有强的配位作用,增强了其对CO2加氢制甲酸的催化性能. 在120 ℃和6 MPa CO2/H2(体积比1∶1)条件下,添加Et3N为有机碱和3 mmol PPh3作为供电子配体,在DMSO和水的混合溶剂中反应2 h,TON可达831. 其催化反应机理如图15所示. 首先,在Et3N的作用下,脱除Cl生成Ru-H中间体. 随后,CO2插入到Ru—H键中. 进一步氢分子与Ru中心配位,在甲酸根的辅助下发生异裂. 甲酸脱除后完成催化循环. 速控步骤是CO2插入Ru—H的过程,配体基团的给电子能力越强则越有利于CO2的插入.
2020年,Mehlana 课题组[54]制备了一种含有Pd(II)活性位点、具有联吡啶二羧酸单元的MOF催化剂Pd@Mg:JMS-2 和Pd@Mn:JMS-2,合成过程如图16 所示. 催化剂使用前需于室温下在甲醇中浸泡24 h 进行活化,得到活化的Pd@Mg:JMS-2a 和Pd@Mn:JMS-2a 催化剂. 活化后的MOF 催化剂在CO2加氢生成甲酸盐时表现出高催化活性. 其中Pd@Mn:JMS-2a催化剂活性最佳,在5 MPa[V(CO2)∶V(H2)=1∶4]和100 ℃条件下,在5 mmol KOH 乙醇溶液中反应24 h,TON 可达9816,TOF 可达409 h−1. 他们提出了该催化剂催化CO2加氢制甲酸的可能反应机理. Pd(II)金属中心作为活性位点,能够活化H2形成Pd二氢化物中间体. 然后从Pd二氢化物中消除HCl,得到活性Pd氢化物. 随后CO2插入到Pd氢化物中生成甲酸根配合物,最后甲酸根解离得到产物,完成催化循环.
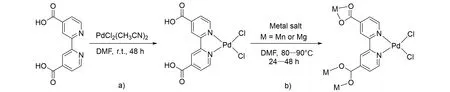
Fig.16 Synthesis of Pd@Mg:JMS⁃2 and Pd@Mn:JMS⁃2[54]
MOF除了被用作催化剂载体,也可用于封装分子催化剂. 2018年,Byers课题组[55]将分子Ru配合物(tBuPNP)Ru(CO)HCl封装到UiO-66空腔中,制备出[Ru]@UiO-66包封催化剂用于CO2加氢生成甲酸盐. 如图17所示,分子Ru催化剂的封装可以通过连接体的解离置换反应进而控制MOF框架开孔关孔实现. 在27 ℃、3 bar(1 bar=0.1 MPa)CO2和12 bar H2的条件下,以DBU 为碱,在DMF 中反应30 min,催化反应的TON 高达30000. 这是当前骨架材料固定分子催化剂达成的最高转化数. 与均相对应物相比,该方法制备的封装催化剂[Ru]@UiO-66更多地保留了分子催化剂的特点,表现出更好的催化活性、更高的可回收性、更慢的失活速度和良好的抗中毒性. 回收实验证明该催化剂具有高稳定性,在连续循环5 次后TON 仍保持不变. 该研究表明,除了在MOFs 表面常规接枝均相催化剂外,通过简单地打开或关闭孔将分子催化剂封装在MOFs 内具有良好的应用潜力.
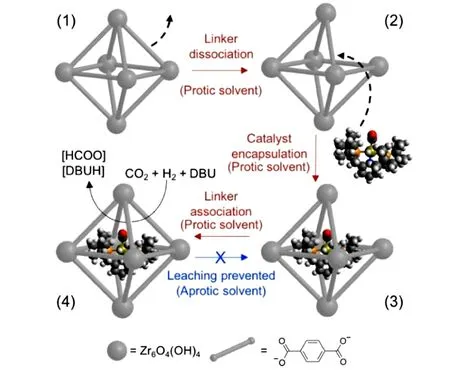
Fig.17 Catalysis in MOFs using aperture⁃opening encapsulation[55]
4 总结与展望
近年来,随着均相分子催化剂在CO2转化到甲酸领域的巨大成功,为解决催化剂的回收利用问题,高效均相催化剂的多相化迫在眉睫. 新兴的多孔骨架材料COFs,POPs和MOFs以其独特的优势成为分子催化剂固定的优良选择. 当前多孔有机骨架材料的合成通常需要较高的反应温度和长的反应时间,有些需要使用复杂的单体和大量的有机溶剂,合成路线繁琐. 拓展不同类型的廉价单体,采用环保便捷的机械化学、超声化学、微波、可见光等方法诱导合成适宜的POPs,COFs和MOFs载体材料是该领域未来研究的一个重要方向.
新型多孔材料固定分子催化剂用于CO2加氢还原转化为甲酸已经取得了一定的成果. 固定的分子催化剂包括常用的均相贵金属Ir、Ru催化剂等以及无金属的路易斯酸碱对催化剂. 催化剂往往以催化性能优良的含膦或氮的双齿配合物或钳型配合物均相催化剂为模型进行设计. 因此当前报道的多孔骨架固定型分子催化剂骨架中多含有双齿或多齿结构的N或P配位点,以此来负载Ir、Ru配合物. 分子催化剂的固定方法包括直接在连接体上配位金属、将分子催化剂接枝到连接体上以及将分子催化剂封装到骨架笼内等. 尽管当前报道的多孔骨架固定型催化剂所需压力、温度条件普遍比较高,令人欣喜的是有些多相催化剂展示出的活性与相应的分子催化剂活性已经非常接近,甚至有所超越. 高活性的催化剂往往存在金属配合物和多孔骨架之间的协调作用,骨架中的N或P位点作为供电子基团增加金属中心的电子密度,从而提升活性. 另外,骨架中丰富的碱性位点有利于吸附CO2,加快反应速率. 这让我们对多孔骨架固定型分子催化剂的发展前景充满期待.
当前,多孔骨架固定分子催化剂的主要问题是与均相催化剂相比活性仍有很大提升空间,无法在温和条件下还原CO2. 此外,分子催化剂结构改变、金属浸出、催化剂重复利用性不佳的问题也亟待解决. 通过开发多孔骨架和分子催化剂的新型连接方式,比如封装方法,有效避免催化反应过程中金属中心解配,有望保持分子催化剂的高活性和稳定性. 另外,精准调控分子催化剂配体的供电子作用以及金属中心与骨架碱性位点的协同作用将有助于开发高活性的多孔骨架催化剂. 未来开发高效、高稳定性的多孔骨架催化剂仍然是该领域面临的巨大挑战.
猜你喜欢
农业资源与环境学报(2022年3期)2022-05-26
核化学与放射化学(2022年2期)2022-04-28
农业资源与环境学报(2021年4期)2021-07-30
无机化学学报(2020年7期)2020-07-20
太原科技大学学报(2020年3期)2020-06-22
钻井液与完井液(2018年2期)2018-06-13
中国蜂业(2018年4期)2018-05-09
科技创新导报(2016年30期)2017-03-15
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
支点(2015年11期)2015-11-16
