
锰改性对ZIF-67 衍生Co3O4 低温催化氧化甲醛性能的影响
2022-08-04 14:53韩小金郑剑锋李巧艳赵青松侯亚芹黄张根
燃料化学学报 2022年7期
向 宁 ,韩小金 ,郑剑锋 ,李巧艳 ,赵青松 ,侯亚芹,* ,黄张根,*
(1. 长治学院生命科学系, 山西 长治 046011;2. 中国科学院山西煤炭化学研究所 煤转化国家重点实验室, 山西 太原 030001;3. 太原理工大学 环境科学与工程学院, 山西 晋中 030600)
甲醛(HCHO)是一种常见的室内空气污染物,主要来自于建筑装饰材料的释放。短期接触甲醛会刺激人体眼睛和上呼吸道,使人咳嗽恶心;长期接触甲醛可能引起肺炎、肺水肿等严重疾病,甚至会导致白血病、鼻咽癌的发生[1−3]。鉴于此,研究人员开发了多种去除甲醛的技术,常见的包括吸附法、等离子体法、光催化法和催化氧化法。其中,催化氧化法由于净化效率高、无二次污染、操作便利,被认为是最理想的甲醛净化技术[4]。根据催化剂活性组分的不同,甲醛氧化催化剂可大致分为贵金属催化剂和过渡金属氧化物两大类[5−8]。2005 年,Zhang 等[9]利用Pt/TiO2催化剂实现了甲醛的室温催化氧化。随后,在此基础上研究人员开发了多种可以实现甲醛室温催化氧化的负载型贵金属催化剂(如Pd/TiO2、Au/CeO2、Ir/TiO2等)[10−14]。然而,贵金属稀少的储量、高昂的价格限制了其大规模推广应用。因此,研究开发廉价易得的高性能甲醛催化氧化材料具有重大的实用价值。
Co3O4良好的氧化还原性能、较低的价格和丰富的储量使其在催化领域广泛应用[15,16]。唐彤等[17]利用Co(NO3)2·6H2O 为钴源、草酸为沉淀剂制备得到的Co3O4催化剂可在140 ℃下实现甲醛的完全氧化。Bai 等[18]利用KIT-6 分子筛作为硬模板制备得到的3D-Co3O4催化剂由于具有较高的比表面积、丰富的Co3+和表面活性氧物种,可在130 ℃下实现甲醛的完全氧化。然而,单一组成的Co3O4催化剂甲醛氧化温度仍旧较高,其低温催化氧化性能仍需要进一步提升。
相比于单一组成的金属氧化物,复合金属氧化物由于元素间的协同作用通常在催化反应中具有更高的催化活性[4,16]。其中,Mn 由于价电子活跃,具有良好的低温氧化还原能力,常作为助剂用于提升催化剂的催化性能[19−21]。Cai 等[22]制备了一系列不同Mn 掺杂量的Co3O4催化剂用于催化氧化1,2-二氯苯。实验结果表明,当Co/Mn 物质的量比为1∶9 时制备得到的催化剂展示了最佳的催化活性,可在347 ℃下实现90% 的1,2-二氯苯转化率。Shi 等[23]研究发现,当MnxCo3−xO4催化剂中Co/Mn 物质的量比为3∶1 时其甲醛氧化活性最佳。然而,由于锰和钴的氢氧化物溶度积(Ksp)不同,采用传统共沉淀法制备的Mn-Co 复合金属氧化物很难实现锰钴物种的均匀分布,从而降低了催化剂的反应性能。另一方面,除了催化剂的组成,催化剂的形貌结构也是影响其催化性能的重要因素。通常来说,大比表面积的催化剂不仅有利于反应物种的吸附,而且有利于活性位点的暴露[4,24]。因此,设计制备比表面积大、组成均匀的Mn-Co 复合金属氧化物催化剂是提升其甲醛氧化活性的有效手段。
近年来,由金属离子/离子簇与有机配体通过配位作用自组装形成的金属有机框架(metal-organic frameworks,MOFs)材料由于其比表面积大、组成可调、结构可控,被认为是一种理想的合成金属氧化物的前驱体[25−27]。Zhao 等[28]通过煅烧不同尺寸的ZIF-67 合成了三种不同粒径的Co3O4催化剂,研究结果表明,小尺寸的Co3O4催化剂由于具有丰富的表面吸附氧和Co3+,在甲苯氧化反应中展示了最佳的催化活性。Xu 等[29]通过煅烧Cu2+部分取代的ZIF-67 得到了双金属CuO/Co3O4催化剂,相比于单一组成的CuO 和Co3O4,双金属CuO/Co3O4催化剂在甲苯氧化反应中展示了更高的催化活性。因此作者推测,如果能够在ZIF-67 中引入锰物种制备Mn 改性的Co3O4催化剂,通过Mn 与Co 之间的相互作用可能会有效提升其甲醛催化氧化性能。目前,双金属ZIF-67 衍生的锰钴复合金属氧化物催化氧化甲醛的研究报道极少。
基于此,本工作通过煅烧单金属ZIF-67 和双金属Mn-ZIF-67 分别制备得到Co3O4和Mn-Co3O4两种催化剂,系统研究了Mn 改性对Co3O4催化剂物理化学性质及甲醛氧化活性的影响,初步探讨了锰钴催化剂结构与甲醛脱除性能之间的构效关系,并利用in-situDRIFTS 技术揭示了甲醛在Mn-Co3O4催化剂上的氧化机理。
1 实验部分
1.1 试剂
实验所用Co(NO3)2·6H2O(AR,99%)、MnCl2·4H2O(AR,99%)、2-甲基咪唑(98%)和CTAB(99%)均购自阿拉丁,实验所用水均为去离子水。
1.2 催化剂的制备
将2 mmol Co(NO3)2·6H2O 溶 于20 mL 去 离 子水中;将9.08 g 2-甲基咪唑、10 mg CTAB 溶于80 mL 去离子水中。上述两溶液混合后在25 ℃下剧烈搅拌20 min,形成的紫色固体经离心、洗涤后置于烘箱中60 ℃干燥12 h,所得样品记为ZIF-67。Mn-ZIF-67 的制备方法同上,只是将2 mmol Co(NO3)2·6H2O 替 换 为1.6 mmol Co(NO3)2·6H2O 和0.4 mmol MnCl2·4H2O(Mn/Co 物 质 的 量 比1∶4)。最后,将干燥后的ZIF-67 和Mn-ZIF-67 置于管式炉中350 ℃焙烧2 h(升温速率1 ℃/min),得到的催化剂分别记为Co3O4和Mn-Co3O4。
1.3 催化剂活性评价
催化剂的甲醛氧化活性评价在自制的固定床反应器中进行。原料气由高纯氮气和高纯氧气混合而成(总N2∶总O2= 4∶1),甲醛和水蒸气分别通过一定流量的氮气吹扫置于恒温水浴中的多聚甲醛和水得到。气体组成为:98.16 mg/m3HCHO,相对湿度(RH)为60%,20% O2,N2为平衡气。总气体流量100 mL/min,催化剂用量0.1 g(40−60 目),空速为60000 mL /(gcat·h)。进出口气体用Gasmet DX4000 型红外气体分析仪在线分析。甲醛转化率计算公式如下:
式 中,[HCHO]in为 进 口 的HCHO 含 量,[CO2]in和[CO2]out分别代表进口和出口的CO2含量。在整个反应过程中,除CO2外没有检测到其他的含碳产物。
1.4 催化剂的表征
X 射线衍射(XRD)测试在Bruker D8 Advance型X 射线衍射仪上进行,射线源为Cu 靶Kα(40 kV,40 mA,λ= 0.15418 nm)。拉曼(Raman)测试在Thermo Scientific DXR 型拉曼光谱仪上进行,激发波长532 nm。氮气吸附-脱附实验在Autosorb-iQ-2 型物理吸附仪上进行,测试前样品在300 ℃下真空脱气6 h。样品的比表面积和孔径分布分别利用BET和BJH 方法计算得到。扫描电镜(SEM)测试在JSM-7500F 型场发射扫描电镜上进行,加速电压10 kV,粉末样品黏到导电胶上喷金处理后放入仪器中进行观察。X 射线光电子能谱(XPS)测试在Escalab 250Xi 型X 射线光电子能谱仪上进行,激发光源为Al 靶Kα(1486.6 eV)。氢气程序升温还原(H2-TPR)和氧气程序升温脱附(O2-TPD)实验在ChemStar TPx 型化学吸附仪上进行,检测器为TCD。H2-TPR测试方法如下:将30 mg 样品置于内径4 mm 的U 型管中,先用Ar(30 mL/min)在300 ℃吹扫样品1 h,冷却至50 ℃后切换为10%H2/Ar 混合气(30 mL/min),待基线稳定后程序升温至800 ℃(10 ℃/min)。O2-TPD 测试方法如下:将100 mg 样品置于内径4 mm的U 型管中,先用Ar(30 mL/min)在300 ℃吹扫样品1 h,冷却至35 ℃后通入3%O2/He 混合气(30 mL/min)吹扫样品1.5 h;紧接着用He(30 mL/min)吹扫样品1 h 除去物理吸附的O2;最后,程序升温至800 ℃(10 ℃/min)。原位漫反射红外(in-situDRIFTS)实验在Bruker Tensor 27 型红外光谱仪上进行。测试前,先将样品在N2气氛下200 ℃处理0.5 h,然后降温至120 ℃采集背景光谱。随后,通入含有甲醛的原料气(98.16 mg/m3)进行反应,采集不同反应时刻的谱图。
2 结果与讨论
2.1 催化剂的性能评价
催化剂的甲醛氧化起燃曲线如图1 所示。由图1 可知,Co3O4催化剂的甲醛氧化活性较低,在90 ℃以下几乎没有甲醛氧化活性(仅5%左右)。随着反应温度的提升,Co3O4催化剂的甲醛转化率逐渐上升,其t50和t90(甲醛转化率达到50% 和90%时所需的温度)分别为118 和129 ℃。相比之下,Mn 掺杂后的Mn-Co3O4催化剂展示了更高的甲醛氧化活性,90 ℃时的甲醛转化率提升至30%左右,并且其t50和t90分别降至104 和118 ℃。上述结果表明,Mn 改性有效提升了Co3O4催化剂的甲醛氧化性能。
图2为Mn-Co3O4催化剂催化氧化甲醛的稳定性测试结果。由图2 可知,在120 ℃条件下,Mn-Co3O4催化剂在48 h 的稳定性测试中始终保持了95%以上的甲醛转化率,表明了其具有良好的稳定性。
2.2 催化剂的表征
2.2.1 XRD 分析
图3所示为催化剂的XRD 谱图。未掺杂的Co3O4催化剂在19.0°、31.3°、36.9°、38.6°、45.0°、55.6°、59.4°和65.4°处的衍射峰分别对应了标准Co3O4(JCPDS 74−2120)的(111)、(220)、(311)、(222)、(400)、(422)、(511)和(440)晶面。而对于Mn-Co3O4催化剂来说,仅观察到属于Co3O4的特征峰,并未检测到与锰物种相关的衍射峰,这可能是由于锰物种处于高度分散的状态或已进入到Co3O4晶格内[23]。值得注意的是,Mn 掺杂后催化剂的衍射峰强度下降,表明Mn 的掺入降低了Co3O4的结晶度。根据Scherrer 公式,利用催化剂(311)晶面分别计算了Co3O4和Mn-Co3O4催化剂的晶粒尺寸,结果见表1。由表1 可知,Mn-Co3O4催化剂的晶粒尺寸更小,而这有助于增大催化剂的比表面积和活性位点的暴露,对于甲醛氧化是有利的[29]。此外,相较于未掺杂的Co3O4催化剂,Mn-Co3O4催化剂(311)晶面衍射峰的2θ值向低角度方向移动(从36.9°偏移至36.7°),这可能是由于离子半径较大的Mn 物种进入到Co3O4晶格内引起晶格膨胀,导致晶格参数增大引起的(表1)[30]。

表1 Co3O4 和Mn-Co3O4 催化剂的结构参数Table 1 Properties of Co3O4 and Mn-Co3O4 catalysts
2.2.2 Raman 分析
图4所示为催化剂的Raman 谱图。未掺杂的Co3O4催化剂在波数为192、472、514、612和678 cm−1处展示了五个Raman 振动峰,分别对应于Co3O4的和A1g模式[22]。相比之下,Mn-Co3O4催化剂的Raman 峰强度有所下降,同时其A1g模式的对称伸缩振动峰从678 cm−1偏移至672 cm−1(红移)。红移现象的出现表明Mn 的掺入导致Co3O4尖晶石结构发生扭曲,降低了化学键强度(力常数),增加了催化剂的缺陷,而这对于O2的吸附与活化是有利的[22,28,29]。
2.2.3 氮气吸附-脱附分析
图5所示为催化剂的氮气吸附-脱附等温线和孔径分布。由图5(a)可知,Co3O4和Mn-Co3O4催化剂均呈现为典型的IV 型等温线,并在相对压力为0.6−1.0 时具有H3 型回滞环,表明两种催化剂中均存在介孔结构且与裂隙孔的形成有关;另外两种催化剂出现回滞环的相对压力较高,根据BJH方法和Kelvin 方程可知其介孔孔径较大,这与图5(b)中的孔径分布结果一致[17,31]。催化剂的比表面积、孔容和平均孔径数据见表1。由表1 可知,Mn 改性增大了催化剂的比表面积和孔容,这与之前XRD推测的结果一致。相比于未掺杂的Co3O4催化剂,Mn-Co3O4催化剂更大的比表面积和孔容不仅有利于反应物分子的吸附,而且有利于活性位点的暴露和表面缺陷的产生,这可能是Mn 掺杂后催化剂甲醛氧化活性提升的原因之一[32,33]。
2.2.4 SEM 表征
图6为催化剂前驱体和催化剂的SEM 照片。从图6(a)、(b)可以看出,ZIF-67 和Mn-ZIF-67 均为标准的立方体结构,其表面光滑密实,平均尺寸约为290 nm。350 ℃煅烧后形成的Co3O4和Mn-Co3O4催化剂基本保持了与其前驱体相同的形貌,但立方体各面收缩且变得粗糙,这可能是由于煅烧过程中配体分解导致的。此外,从Mn-Co3O4催化剂的EDS-mapping 图上可以看出,Mn、Co、O 元素在催化剂上均匀分布,进一步表明Mn 物种已经成功地引入到Co3O4催化剂中。
2.2.5 XPS 分析
图7(a)−(c)分别为催化剂的Co 2p、Mn 2p和O 1s谱图。如图7(a)所示,Co3O4催化剂在779.56和794.63 eV 处的峰分别归属于Co 2p3/2和Co 2p1/2光谱峰。Mn 改性后,催化剂的Co 2p3/2结合能从779.56 eV 移动至780.09 eV,这可能是由于Co 物种的部分电子转移至Mn 物种上使Co 物种的电子密度减少导致的,该结果表明,Mn-Co 间存在强相互作用,而这对于增加催化剂表面缺陷含量是有利的[19,32]。两催化剂的Co 2p分裂能级差(ΔECo2p=ECo2p1/2−ECo2p3/2)均为15.10 eV 左右,表明催化剂中Co2+和Co3+共存[33]。拟合结果(表2)显示Mn-Co3O4催化剂具有更高比例的Co3+,这进一步证明了Mn改性会降低Co 物种的电子密度,而Co3+强的氧化活性有利于提升催化剂的甲醛氧化性能[4]。
图7(b)为Mn-Co3O4催化剂的Mn 2p谱图。其中,Mn 2p3/2光谱峰可以拟合为Mn2+(640.70 eV)、Mn3+(642.00 eV)和Mn4+(643.80 eV)三个峰。由表2可知,Mn-Co3O4催化剂中Mn4+/Mn 比值约为0.43,表明其具有丰富的Mn4+物种。文献报道,充足的Mn4+物种有助于增强催化剂的氧化还原循环,对于甲醛氧化反应是有利的[24,30]。
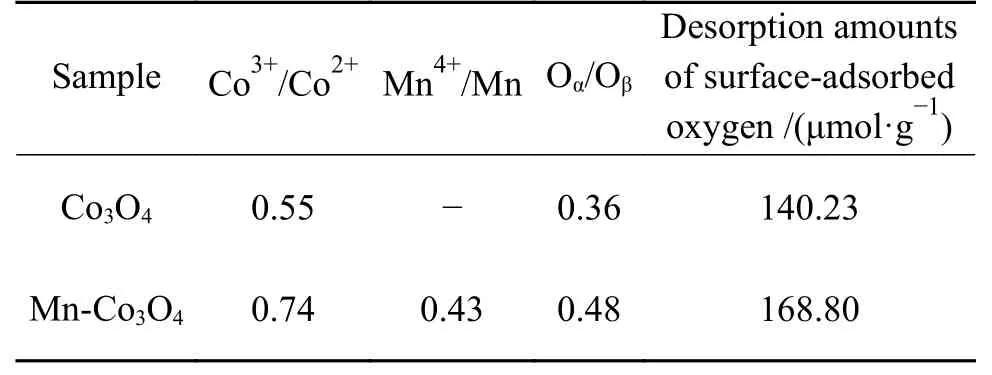
表2 Co3O4 和Mn-Co3O4 催化剂的XPS 表征和表面吸附氧脱附量Table 2 XPS results and desorption amounts of surface-adsorbed oxygen of Co3O4 and Mn-Co3O4 catalysts
图7(c)为催化剂的O 1s谱图。其中,位于529.90−530.27 eV 和531.21−531.35 eV 处的峰分别归属于晶格氧(Oβ)和表面吸附氧(Oα)物种。相比于Co3O4催化剂,Mn 改性后的Mn-Co3O4催化剂其晶格氧结合能向高能方向移动,这可能是由于Mn-Co 间的强相互作用降低了催化剂的化学键强度,使晶格氧流动性增强、电子密度降低导致的,这与Raman结果一致[34−36]。此外,由表2 可知,Mn-Co3O4催化剂上具有更高比例的表面吸附氧物种,而表面吸附氧物种被认为是甲醛催化氧化过程中的活性氧物种[37]。因此,表面吸附氧物种比例更高的Mn-Co3O4催化剂展示了更好的甲醛氧化活性。
2.2.6 H2-TPR 分析
图8所示为催化剂的H2-TPR 谱图。由图8 可知,Co3O4催化剂展示了两个连续的还原峰,其中,在315 ℃处的肩峰和386 ℃处的主峰分别归属于Co3+→Co2+和Co2+→Co0的 还 原[18]。与Co3O4催化剂的还原过程类似,Mn 掺杂后的Mn-Co3O4催化剂同样展示了两个连续的还原峰,并没有出现额外的还原峰,表明Co 物种还原的过程伴随着Mn 物种的还原。根据文献[30]可知,第一个还原峰归属于Co3+→Co2+和Mn4+→Mn3+的还原;第二个还原峰归属于Co2+→Co0和Mn3+→Mn2+的还原。值得注意的是,Mn 物种引入后催化剂初始还原峰向低温方向移动,表明Mn 物种的引入提升了催化剂的低温氧化还原能力,而这对于甲醛催化氧化反应是有利的。
2.2.7 O2-TPD 分析
图9所示为催化剂的O2-TPD 谱图。从图9 中可以看出,Co3O4和Mn-Co3O4催化剂的氧脱附峰大致可以分为三个区域,其中,< 400 ℃的峰归属于表面吸附氧物种;400−700 ℃处的峰归属于表面晶格氧物种;> 700 ℃的峰归属于体相晶格氧物种[18,37]。相比于Co3O4催化剂,Mn-Co3O4催化剂具有更丰富的表面吸附氧物种(168.80 vs 140.23 μmol/g),这与前文的XPS 结果一致。该结果表明,Mn-Co3O4催化剂具有更强的氧活化能力,这可能是由于Mn-Co3O4催化剂更大的比表面积,更低的结晶度以及Mn-Co 间的强相互作用促进了活性位点的暴露,增加了催化剂的缺陷,从而促进了氧气在Mn-Co3O4催化剂上的吸附与活化。此外,Mn-Co3O4催化剂表面晶格氧脱附温度更低(从580 ℃降低至562 ℃),表明Mn 掺杂提升了晶格氧的流动性,这进一步验证了Raman 和XPS 的结果。
2.2.8In-situDRIFTS 研究
图10为120 ℃下Mn-Co3O4催化剂通入甲醛原料气后的原位红外光谱谱图。从图10 可以看出,在整个反应过程中始终未检测到吸附态甲醛分子的红外吸收峰,表明甲醛可以在Mn-Co3O4催化剂上迅速氧化。随着反应时间的延长,分别在3600−3100、2930、1560、1540、1440、1340和1318 cm−1处检测到了红外吸收峰。其中,3600−3100 cm−1处的宽峰为水分子中O−H 的伸缩振动[11];1440 cm−1处的峰归属于亚甲二氧基(DOM)物种的OCO 伸缩振动[38];2930 cm−1处的峰归属于甲酸盐物种(HCOO−)的C−H 反对称伸缩振动[18];1560 和1540 cm−1处的峰归属于甲酸盐物种的COO 反对称伸缩振动[37,38];1340 和1318 cm−1处的峰为甲酸盐物种的COO 对称伸缩振动[9,39]。因此,亚甲二氧基和甲酸盐物种是甲醛在Mn-Co3O4催化剂上催化氧化的主要中间物种。
最后,基于原位红外光谱结果推测了甲醛在Mn-Co3O4催化剂上可能的氧化路径,如图11 所示。首先,O2在Mn-Co3O4催化剂上解离活化形成表面吸附氧物种。随后,表面吸附氧物种与HCHO分子发生亲核取代形成亚甲二氧基(DOM)物种。DOM 物种进一步转化成为甲酸盐(HCOO−)物种。最终,甲酸盐物种被表面吸附氧物种氧化形成CO2和H2O,完成催化循环。
3 结 论
通过煅烧单金属ZIF-67 和双金属Mn-ZIF-67分别制备得到Co3O4和Mn-Co3O4两种催化剂,系统研究了Mn 改性对Co3O4催化剂甲醛氧化性能和物化性质的影响。相比于未改性的Co3O4催化剂,Mn 改性后的Mn-Co3O4催化剂甲醛氧化活性显著提升,其t90从129 ℃下降至118 ℃。表征结果显示,Mn 改性后催化剂的结晶度降低,缺陷和比表面积增加,这有利于反应物分子的吸附和活性位点的暴露;同时,Mn-Co 间存在的强相互作用显著改善了Mn-Co3O4催化剂的低温氧化还原性能和氧活化能力,使其具有更加丰富的Co3+和表面吸附氧物种。最终,这些因素共同促进了Mn-Co3O4催化剂对甲醛的降解。此外,in-situDRIFTS结果表明,亚甲二氧基和甲酸盐物种是甲醛在Mn-Co3O4催化剂上催化氧化的主要中间物种。
猜你喜欢
佛山科学技术学院学报(自然科学版)(2022年5期)2022-10-09
物理学报(2022年15期)2022-08-12
光子学报(2022年6期)2022-07-27
小学生学习指导(高年级)(2022年3期)2022-03-29
当代作家(2021年11期)2021-12-17
作文周刊·八年级版(2020年8期)2020-05-25
黄河黄土黄种人(2019年11期)2019-12-20
小学生学习指导(高年级)(2019年4期)2019-11-27
健康博览(2019年1期)2019-04-08
小学生导刊(高年级)(2017年2期)2017-06-10
