
HFAU 分子筛非骨架铝物种对Brønsted 酸性质影响的理论研究
2022-08-04 14:53秦玉才宋丽娟
燃料化学学报 2022年7期
郑 健 ,李 强 ,秦玉才 ,宋丽娟,*
(1. 中国石油大学(华东) 化学工程学院, 山东 青岛 266555;2. 辽宁石油化工大学 石油化工催化科学与技术重点实验室, 辽宁 抚顺 113001)
FAU 型分子筛作为一种酸催化剂被广泛应用于催化裂化、加氢裂化等化工过程中,而Brønsted酸中心(简称B 酸)的性质是影响其应用性能的关键因素。几十年来,人们一直关注骨架和非骨架物种对Brønsted 酸性质的影响[1−8]。其中,分子筛改性和处理过程形成的非骨架铝物种与B 酸中心的内在联系一直是人们关注的热点[9−13]。
大量研究表明,非骨架铝物种的存在不仅改善了分子筛的热稳定性,而且提高了分子筛的催化活性。Carvajal 等[14]在FAU 分子筛上研究了正己烷的裂化反应,他们发现不论硅铝比是多少,含有非骨架铝(Extra-Framework Al,EFAl)物种的分子筛都显示更好的活性。Wang 等[15]发现分子筛中正庚烷的裂化活性会随着非骨架铝物种数量的减少而降低。Mota 课题组[16]发现,有EFAl 存在的USY 分子筛3-甲基戊烷的氢氘交换速率提高了20 多倍。因此,非骨架铝物种对FAU 分子筛催化性能影响的机理研究对催化剂的开发具有很强的指导意义。
刘中民课题组[17]通过固体核磁共振方法研究了脱铝FAU 分子筛中非骨架铝物种对B 酸酸性的影响,结果表明,非骨架铝物种的出现提高了B酸中心的酸强度。文献[15,18,19]同样采用核磁技术证明了四配位非骨架铝物种的形成增强了B 酸的酸强度,提高了烷烃的裂化活性。Mota 研究组[20,21]认为,非骨架铝物种通过氢键稳定分子筛的共轭碱位从而提高了B 酸中心的酸强度。而Li 等[22]则认为B 酸强度的增强是非骨架铝物种与骨架铝相邻氧原子配位成键,降低了B 酸中心质子的亲和力。因此,相邻非骨架铝物种的影响机制普遍认为是通过极化作用增强了分子筛的B 酸强度,进而提高了分子筛的催化活性。
另外,有一些其他的意见解释非骨架铝物种提高分子筛催化活性的原因。Van Bokhoven 等[23,24]发现分子筛经过蒸汽处理前后B 酸具有相似的酸强度,他们认为裂化速率的提高与烷烃吸附热的增加有关,这将导致沸石中反应物的浓度增加。Gounder研究小组[25]认为,非骨架铝物种的促进作用主要与HFAU 中有效孔隙尺寸减小有关, 可以通过色散作用更好的稳定过渡态结构,从而提高了裂化活性。Schallmoser 等[12]提出含非骨架铝物种的分子筛正戊烷裂化活性的提高主要归因于B 酸中心周围微环境的变化而非极化作用。上述论述表明非骨架铝物种的存在与分子筛活性的增加有关,但非骨架铝与B 酸的协同作用机制仍然没有明晰。
合理探针分子(烷烃、烯烃、丙酮、氨、吡啶和苯等)的选择对于了解分子筛B 酸位点的吸附性能具有重要意义。噻吩分子具有弱碱性,可与质子结合研究分子与酸性位点的作用模式。且与苯相似,噻吩有可产生亲电取代反应的芳香性。因此,噻吩也是一种非常有代表性的探针分子,研究多个活性位点间的协同作用模式。
本研究主要以不同的非骨架铝物种[Al(OH)2+和Al3+]为研究对象,以噻吩为探针分子,探索非骨架铝物种(Extra-Framework Al,EFAl) 对B 酸性质的影响。从量子力学角度考察了活性位微环境及其与噻吩间的关联性。此工作可以为未来高效催化剂中有效物种活性位的构建提供新的思路和强有力的理论支撑。
1 模型搭建与计算方法
1.1 模型搭建
作者搭建了具有Fd-3 对称性的三斜菱状周期性FAU 分子筛单位胞模型,基本参数为a=b=c=17.2 Å、α=β=γ= 60.0°,组成为Si48O96。以此模型为基础搭建了含非骨架铝物种的HFAU 模型。EFAl 物种的结构和位置是根据以前的文献报道构建的。许多研究表明,具有羟基结构的四配位非骨架物种是分子筛体系首选的Lewis 酸位点[11,12,26]。Liu 等[11]研究单核EFAl 物种的转化过程时发现Al(OH)2+非骨架铝物种容易脱水形成Al(OH)2+非骨架铝物种,认为Al(OH)2+非骨架铝物种为最优的单核非骨架铝物种。Yi 等[26]认为Al3+非骨架铝物种因其强Lewis 酸性对提高催化剂活性深具潜力。Liu 等[11]在研究多核非骨架铝物种形成的过程中发现,多核非骨架铝物种稳定存在于FAU 分子的β笼中,本研究是在FAU 分子筛超笼中进行的,因此,只考察单核非骨架铝物种。根据Liu 等[11,12,26]对分子筛中非骨架铝物种的研究,选择四配位Al(OH)2+和三配位Al3+作为典型的单核非骨架铝物种。此外还考虑了Al(OH)2+羟基物种的两个方向对B 酸性质的影响。记此模型为1-HFAU/Al(OH)2+, 2-HFAU/Al(OH)2+和HFAU/Al3+。为了后续研究分子筛本征特性做准备,对1-HFAU/Al(OH)2+, 2-HFAU/Al(OH)2+和HFAU/Al3+分子筛模型中的硅铝桥羟基(即B酸位)原子和非骨架铝(Al(OH)2+和Al3+)物种(即L 酸位)原子进行了编号。优化后的结构如图1所示。
1.2 计算方法
本研究应用美国Accelrys 公司的Materials Studio 5.5 软件包中的Dmol3 模块完成,使用广义梯度近似(GGA)[27,28]中的PBE[29]作为电子交换相关函数,采用可极化的双数值基组(DNP),对原子中心电子采用DFT semi-core Pseudopotential (DSPP)进行处理。电子自洽计算收敛至1 × 10−5Ha,为了加速收敛过程,thermal smearing 值设为0.002 Ha。
去质子化能:本研究利用去质子化能(deprotonation energies,DPE)作为表征分子筛酸性的一个重要参数,它表示从团簇中去除酸性质子所需的能量,其定义如下:
式中,EZH为中性分子筛体系的能量,为去掉酸质子后分子筛体系的能量,为气相H 原子的能量。DPE的值不依赖质子接受体系,提供一种独立于反应或吸附分子的酸强度标度,去质子化能越小,表明酸性越强。
吸附能:不同非骨架铝物种的影响下,吸附质分子在分子筛中酸性位的吸附能Eads定义如下:
式中,Eadsorbent为分子筛模型体系的能量,Eadsorbate为吸附质噻吩分子的能量,Eadsorbent-adsorbate为模型化合物在分子筛模型上吸附后的体系总能量。所得吸附能Eads为负值,说明吸附为放热过程,且吸附能的绝对值越大,说明吸附越稳定。
由于电子数N是不连续的,因而式(3)无法直接计算,对式(3)分别取左右极限(即左右导数)及两者的平均值,利于有限差分近似得到三种不同的反应指数:
式中,ρN、ρN+1和ρN−1分别代表体系原始状态(N电子)、结合一个电子状态(N+1 电子)和电离掉一个电子状态(N−1 电子)下的电子密度。
2 结果与讨论
2.1 分子筛活性位和噻吩分子的本征特性
1-HFAU/Al(OH)2+、2-HFAU/Al(OH)2+和HFAU/Al3+分子筛Brønsted 酸(简称B 酸)活性位(Si7O7(H7)Al7)和Lewis 酸(简称L 酸)活性位(AlEF(OH)2+和AlEF3+)的结构和电荷性质如图2 和表1 所示。从图表可以清晰看出,三种分子筛B 酸位Si7、O7、Al7和H7的Mulliken电荷均为2.33、−1.11、2.08和0.55。结果表明,同一种非骨架物种不同羟基方向及不同非骨架铝物种对B 酸的电荷性质没有影响。两个方向Al(OH)2+非骨架铝物种AlEF、O 和H 原子的电荷 分别 为1.93、(−1.12) − (−1.13)和0.51,Al3+非骨架铝物种AlEF的电荷2.02。研究发现,两个方向的羟基非骨架物种(Al(OH)2+)具有相似的电荷性质,Al3+非骨架铝物种中Al 原子的正电荷高于Al(OH)2+非骨架铝物种中Al 原子(2.02 vs 1.93),证明Al3+非骨架铝物种的L 酸强度高于Al(OH)2+非骨架铝物种。此外非骨架铝落位的六元环上Si和Al 原子的正电荷为2.21−2.25 和2.02−2.03,Al原子电荷性质相似,硅原子略有差异主要归因于两种非骨架铝物种的正电荷差异,即L 酸强度差异。六元环上的骨架氧原子主要分为与Si 和Al相连的Si–O–Al 和与Si 和Si 相连的Si–O–Si,其负电荷分别为(−1.13) − (−1.14)和(−1.18) − (−1.21),即随着骨架Al 原子数目的增多骨架氧原子的负电荷增加,碱性增强。
去质子化能是表征分子筛B 酸强度的重要参数,表示从体系中去除酸性质子所需的能量,去质子化能越大,质子给予的能力越弱,B 酸强度就越弱;相反,去质子化能越小,质子给予的能力越强,B 酸强度就越强。由图3 可知,1-HFAU/Al(OH)2+、2-HFAU/Al(OH)2+和HFAU/Al3+分子筛B 酸位的去质子化能分别为1035、1047 和1056 kJ/mol,结果表明,三种分子筛B 酸位具有相似的酸强度(1045 ±11)kJ/mol,与文献结果一致[31]。
图4给出了噻吩分子的福井函数图,图4(a)为福井函数(−),即Fukui(−)表示噻吩分子的亲电进攻指数,红色区域(正值大)亲电进攻指数高,容易贡献电子,蓝色区域(正值小)亲电进攻指数低,不易贡献电子。图4(a)可以看出,噻吩的C 原子亲电进攻指数高,容易贡献电子。图4(b)为福井函数(+),即Fukui(+),表示噻吩分子的亲核进攻指数,红色区域(正值大)亲核进攻指数高,容易接收电子,蓝色区域(正值小)亲核进攻指数低,不易接收电子。由图4(b)可以看出,噻吩的H 原子和S 原子亲核指数高,容易接收电子。由于酸性位点与容易贡献电子的原子作用,因此,酸性位点会优先与噻吩的C 原子作用。
重要的分子轨道是决定一个体系反应发生的关键,分子的最高占据轨道(HOMO)对其电子的束缚较为松弛,具有电子给予体的性质,而最低未占轨道(LUMO) 则对电子的亲和力较强,具有电子接受体的性质,这两种轨道最易互相作用,在化学反应过程中起着极其重要作用。为了进一步研究分子筛的电子性质,分析了1-HFAU/Al(OH)2+、2-HFAU/Al(OH)2+和HFAU/Al3+分子筛中包括前线轨道在内的几个重要分子轨道(LUMO、LUMO-1和LUMO-2),如图5 所示。图5(a)和5(b)是1-HFAU/Al(OH)2+分子筛的LUMO 和LUMO-1,从图可知,LUMO 在L 酸位(Al(OH)2+)上呈现高电子密度,LUMO-1 在B 酸位呈现高电子密度。由此可知1-HFAU/Al(OH)2+分子筛非骨架物种Al(OH)2+比B 酸位更容易接受电子。图5(c)和5(d)是2-HFAU/Al(OH)2+分子筛的LUMO和LUMO-1,其轨道高电子密度区域分别分布在L 酸位(Al(OH)2+) 和B 酸位,与1-HFAU/Al(OH)2+分子筛相似。由此可得非骨架铝羟基的方向不影响分子筛酸性位的电子性质。不同的是HFAU/Al3+分子筛,其LUMO 和LUMO-1 轨道的高电子密度区域均分布在L 酸位,LUMO-2 轨道的高电子密度区域分布在B 酸位,由此可知,相比Al(OH)2+非骨架铝物种,Al3+非骨架铝物种与B 酸位接受电子能力的差异更大,这可能归因于Al3+非骨架铝物种更强的L 酸强度(参考表1)。
分子筛中重要分子轨道的能量和噻吩分子HOMO 的能量如图6 所示,从图6 可以看出,噻吩分子HOMO 能量为−5.755 eV,1-HFAU/Al(OH)2+分子筛LUMO 和LUMO-1 能量为−2.247 和−1.672 eV,与噻吩HOMO 的能量差为3.508 和4.083 eV。2-HFAU/Al(OH)2+分 子 筛LUMO 和LUMO-1 能 量 为−2.244 和−1.617 eV,与 噻 吩HOMO 的 能 量 差为3.511 和4.138 eV。上述结果表明,含不同羟基方向的Al(OH)2+非骨架铝物种分子筛具有相似的轨道能量。对于HFAU/Al3+分子筛其LUMO、LUMO-1和LUMO-2 能量为−3.849、−2.496 和−1.734 eV,与噻 吩HOMO 能 量 差 为1.906、3.259 和4.021 eV。结果表明,含Al3+非骨架物种分子筛LUMO 与噻吩分子的HOMO 具有最低的能量差,最容易与噻吩作用,而且与Al(OH)2+非骨架物种相比具有绝对的优势。
2.2 噻吩在酸性位的吸附
图7给出了噻吩在HFAU/Al3+分子筛L 酸位的吸附能和吸附构型。图7(a)和图7(b)是噻吩在L 酸位吸附的两种构型,分别为噻吩C 原子与Al3+的作用(记为C 吸附模式)和S 原子与Al3+的作用(记为S 吸附模式),对应的吸附能分别为−398 和−367 kJ/mol。 结果表明,噻吩与Al3+位具有强的吸附作用,归因于非骨架铝Al3+的强L 酸性,与Al3+的电荷性质和轨道性质分析结果一致。此外,C 吸附模式的吸附能明显高于S 吸附模式,结果与噻吩的福井函数结果一致,C 具有更强的贡献电子能力。
噻吩在HFAU/Al3+分子筛Al3+位点吸附的差分电子密度图如图8 所示,图中蓝色区域代表电子密度增加的区域,紫色区域则代表电子密度减小的区域,TP 是thiophene 的缩写这里指噻吩。从图中可以看出,C 吸附模式和S 吸附模式的电子主要集中在C 与Al3+物种之间和S 与Al3+物种之间,再结合Al3+与骨架氧的电子分布可以得出C 与Al3+之间的作用强于S 与Al3+之间的作用。两种作用模式的差异必然会导致噻吩分子本身电子重排的差异,图7(a)可以看出,电子偏向与Al3+作用的C–C键,使电子分布对称性发生变化。相比之下图7(a)的电子分布基本保持了原本的C2v对称性。电子重排的差异会进一步影响噻吩分子的活化。
噻吩在HFAU/Al3+分子筛Al3+位点吸附前后C–C 键和C–S 键的变化列于表2。从表2 可以看出,噻吩以C 模式吸附在Al3+位点时C3–C4 键长增加,从0.138 nm 增加到0.142 nm。C4–S 键长减小,从0.172 nm 减小到0.169 nm(编号参考图7)。结果表明噻吩的C3–C4 键被活化了,从偏向双键向单键转变,打破了噻吩分子原来的C2V对称性,噻吩以S 模式吸附在Al3+位点时C4–S 和C1–S 增加,均 从0.172 nm 增 加 到0.176 nm,C1–C2 和C3–C4 键长减小,均从0.138 nm 减小到0.136 nm。结果表明,噻吩的S–H 键被活化了,且键长的变化基本维持了原来的C2V对称性。结果与上述电子差分密度结果一致。
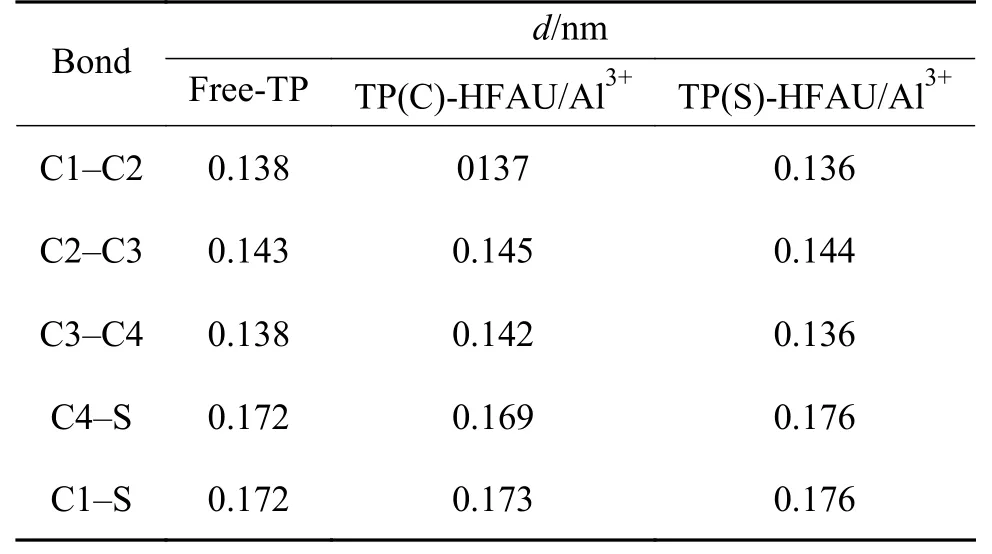
表2 噻吩在HFAU/Al3+分子筛Al3+位点吸附前后C–C 键和C–S 键的变化Table 2 Changes of C–C and C–S bonds of thiophene adsorbed on Al3+ site in HFAU/Al3+ zeolite
噻吩在HFAU/Al(OH)2+分子筛Al(OH)2+位点的吸附构象见图9,图9(a)和9(d)是噻吩在Lewis 酸位(Al(OH)2+)吸附的初猜结构,优化后噻吩会吸附在B 酸位上,吸附第二个噻吩分子时才会吸附在L 酸 (Al(OH)2+) 物种上,而且吸附在Al(OH)2+物种的羟基上并非Al 原子上,这与Al(OH)2+物种的结构和配位有关。上述结果表明,含Al(OH)2+非骨架铝物种分子筛中噻吩会优先吸附在B 酸位。这与轨道性质的分析结果有一点差异,尽管相比B 酸位,Al(OH)2+物种更容易接受电子,但是由于Al(OH)2+物种本身结构的影响,使噻吩更容易在B 酸位吸附,这说明主体电子性质和结构性质均是决定分子吸附模式的关键。
噻吩在HFAU/Al(OH)2+分子筛B 酸和L 酸(Al(OH)2+)位点的吸附能如图10 所示,噻吩在1-HFAU/Al(OH)2+和2-HFAU/Al(OH)2+分子筛B 酸位的吸附能分别为−158 和−141 kJ/mol。虽然两分子筛酸性位具有相似的电子性质(参考本文2.1 节)但吸附能相差17 kJ/mol,造成吸附能差异的原因是Al(OH)2+物种对噻吩分子施加一个弱的相互作用。因此,Al(OH)2+羟基方向不同,作用不同,吸附能不同。噻吩分子在1-HFAU/Al(OH)2+和2-HFAU/Al(OH)2+分子筛Al(OH)2+位上的吸附能分别为−290和−146 kJ/mol,吸附能差值相差144 kJ/mol,结合图9(c)和9(f)分析发现,1-HFAU/Al(OH)2+分子筛Al(OH)2+物种羟基的方向发生变化了,且Al(OH)2+物种中的骨架氧原子向着B 酸位方向偏移,与吸附在B 酸位的噻吩作用,增加了噻吩的吸附能。
2.3 酸性位之间的协同作用模式
为了进一步研究非骨架铝物种与B 酸位之间的协同作用机制,计算了噻吩在1-HFAU/Al(OH)2+、2-HFAU/Al(OH)2+和HFAU/Al3+分子筛B 酸位点的吸附,其吸附构型和差分电子密度如图11 所示。图11(a) 中Al(OH)2+的H 原子与吸附在B 酸位噻吩分子S 原子作用。图11(b)中Al(OH)2+的O 原子与吸附在B 酸位噻吩分子H 原子作用。图11(c)由于Al3+的结构原因几乎对吸附在B 酸位噻吩分子没有影响。差分电子密度图中可以看出图11(e)中的Al(OH)2+非骨架铝物种与噻吩的作用最显著,其次是图11(d)中的Al(OH)2+非骨架铝物种,而Al3+非骨架铝物种与噻吩分子没有作用。图12 给出了噻吩在1-HFAU/Al(OH)2+、2-HFAU/Al(OH)2+和HFAU/Al3+分子筛B 酸位的吸附能,分别为−175、−218 和−140 kJ/mol。结果发现,与非骨架铝物种Al3+相比,非骨架铝物种Al(OH)2+与噻吩分子的弱相互作用显著增加了噻吩的吸附能,证明了分子筛中弱相互作用对稳定分子的重要性,且从电子层面揭示了非骨架铝物种与B 酸位之间的协同作用模式。
3 结 论
本研究采用周期性密度泛函理论的方法研究了典型四配位和三配位单核非骨架铝物种Al(OH)2+和Al3+对Brønsted 酸(简称B 酸)性质的影响。通过结构、电荷和轨道性质分析可以发现非骨架铝物种(Al3+)中Al 原子的正电荷高于非骨架铝物种(Al(OH)2+)中Al 原子的正电荷(2.02 vs 1.93)。HFAU/Al(OH)2+和HFAU/Al3+分子筛最低未占轨道(LUMO)在Al(OH)2+和Al3+物种呈高电子密度,HFAU/Al3+分 子 筛LUMO 具 有 最 低 的 轨 道 能 量(−2.2 vs−3.8 eV )。证明Al3+非骨架铝物种的Lewis 酸(简称L 酸)强度高于Al(OH)2+非骨架铝物种。电子性质结合去质子化能的结果发现,1-HFAU/Al(OH)2+、2-HFAU/Al(OH)2+和HFAU/Al3+分子筛B 酸位具有相 似 的 酸 强 度(1045 ± 11)kJ/mol。由 此 可 得,Al(OH)2+和Al3+非骨架铝物种羟基方向的差异和L 酸强度的差异不影响B 酸中心的酸强度。通过模拟噻吩分子的吸附,获得吸附能及吸附过程中电子性质、几何结构的变化。结果发现,HFAU/Al3+分子筛中噻吩易于吸附在Al3+位,归因于Al3+中心的强L 酸性。而HFAU/Al(OH)2+分子筛中的噻吩会优先吸附在B 酸位而非Al(OH)2+活性中心,归因于Al(OH)2+物种的结构特性。此外,Al(OH)2+非骨架铝物种可以对吸附在B 酸位的噻吩施加一个弱的相互作用(色散作用)促进噻吩的吸附。吸附的模式依赖Al(OH)2+物种的结构和羟基的方向。此项工作从电子层面探究了主体HFAU 分子筛(含非骨架铝)和客体噻吩分子的本征特性,揭示了活性位间的协同作用模式,为高效催化剂和吸附剂的设计提供理论指导。
猜你喜欢
山西大同大学学报(自然科学版)(2022年3期)2022-07-06
电子乐园·上旬刊(2022年5期)2022-04-09
黑龙江大学自然科学学报(2022年1期)2022-03-29
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
煤气与热力(2021年9期)2021-11-06
中国新技术新产品(2020年5期)2020-05-06
农业工程技术·温室园艺(2017年3期)2017-07-13
汽车零部件(2016年6期)2016-07-18
