
桂利嗪过饱和自乳化释药系统的制备与质量评价
2022-08-03 01:47刘挺峰
西北药学杂志 2022年5期
刘挺峰
武汉市红十字会医院,武汉 430015
桂利嗪(cinnarizine,CIN)属于哌嗪类钙拮抗剂,主要用于治疗中风、脑动脉硬化等脑血管疾病[1]。CIN 属于生物药剂学分类系统(BCS)中的Ⅱ类弱碱性药物,其溶解度具有pH 依赖性,其在pH 6.8介质中的溶解度为2.2 μg·mL-1,在pH 3介质中溶解度为52.3 μg·mL-1[2],表现为较差的生物利用度以及较大的个体差异性[3]。自乳化释药系统(self-emulsifying drug delivery system,SEDDS)是一种改善难溶性药物口服生物利用度的有效策略[4],但是对于载有弱碱性药物的SEDDS而言,其经胃液稀释后,溶解在胃液中的药物进入肠道中会析出沉淀[5],影响药物吸收,而在SEDDS处方中加入沉淀抑制剂可降低药物在胃肠道中发生沉淀的风险[6]。本研究将桂利嗪制备成自乳化释药系统,并加入沉淀抑制剂[7],抑制药物析出沉淀,有望进一步提高CIN的口服生物利用度。
1 仪器与试药
1.1 仪器
ZNCL-BS-X19型数显磁力搅拌器(河南爱博特科技发展有限公司);Zetasizer Nano ZS 90型激光粒度仪(英国马尔文公司);TG18G 型高速离心机(上海赫田科学仪器有限公司);JEM-2100F 型透射电镜(日本电子株式会社);RT600型溶出度仪(深圳市锐拓仪器设备有限公司);SYC-A 型数显水浴恒温振荡器(常州金坛精达仪器制造有限公司)。
1.2 试药
CIN(批号20200818,天津天药药业股份有限公司);蓖麻油(castor oil)、聚乙二醇(PEG 400)均购自上海化学试剂公司;聚山梨醇酯20(Tween 20)、聚山梨醇酯80(Tween 80)均购自南京威尔化工有限公司;聚氧乙烯蓖麻油(cremophor EL 35)、聚氧乙烯氢化蓖麻油(cremophor RH 40)、聚乙二醇硬脂酸酯(solutol HS 15)、辛酸/癸酸三酰甘油(miglyol 812)、泊洛沙姆127(poloxamer 127)和聚乙烯己内酰胺-聚醋酸乙烯酯-聚乙二醇接枝共聚物(soluplus)均由巴斯夫应用化工有限公司惠赠;油酸乙酯(ethyl oleate)和油酸(oleic acid)均由国药集团化学试剂公司惠赠;单油酸甘油酯(peceol)、月桂酸聚乙二醇甘油酯(gelucire 44/14)、聚氧乙烯-8-辛酸/癸酸酯(labrasol)、单亚油酸甘油脂(maisine35-1)、丙二醇单辛酸酯(capryol 90)、肉豆蔻酸异丙酯(isopropyl myristate,IPM)、中链三酰甘油(medium chain triglycerides,MCT)、二乙二醇单乙基醚(transcutol HP)、单辛酸甘油酯(capmul MCM C8)均由嘉法狮贸易有限公司惠赠;甲醇为色谱纯;其他试剂均为分析纯;桂利嗪片(广东华南药业集团有限公司)。
2 方法与结果
2.1 CIN 平衡溶解度
将油相组分(蓖麻油、油酸、油酸乙酯、miglyol 812、peceol、maisine 35-1、capryol 90、MCT、IPM 和capmul MCM C8)、表面活性剂(Tween 20、Tween 80、cremophor EL 35、cremophor RH 40、solutol HS 15、labrasol 和gelucire 44/14)和助表 面活性 剂(transcutol HP和PEG 400)分别加入含有过量CIN原料药的棕色玻璃瓶中,涡旋混合5 min,随后将混合物放置在37 ℃水浴中,以100 r·min-1的转速摇动48 h,使药物溶解达到平衡。将混合物以6 000 r·min-1离心10 min,取上清液经0.22 μm 滤膜过滤,续滤液经甲醇适当稀释后进样检测药物含量,计算药物的平衡溶解度。结果见表1。
表1 桂利嗪在不同辅料中的溶解度 (n=3,)Tab.1 Solubility of CIN in various excipients (n=3,)

表1 桂利嗪在不同辅料中的溶解度 (n=3,)Tab.1 Solubility of CIN in various excipients (n=3,)
SEDDS中的药物应呈完全溶解的状态,因此优先选择对药物溶解度较高的辅料用于SEDDS的开发[8]。由表1可知,与其他油相比较,药物在油酸和MCT 中的溶解度较高,均大于80 mg·g-1;表面活性剂Tween 20和Tween 80对药物的溶解性较好;助表面活性剂transcutol HP 对药物表现出良好的溶解性。最终选择油酸和MCT 作为油相,Tween 20和Tween 80作为表面活性剂,transcutol HP作为助表面活性剂,通过伪三元相图进一步确定CIN-SEDDS的处方组成。
2.2 伪三元相图
伪三元相图是研究SEDDS处方的常用工具,通过水滴定法构建伪三元相图以确定最佳自乳化区域[9]。将筛选得到的油相、表面活性剂和助表面活性剂按照一定质量比混合,涡旋获得透明溶液。取上述混合液0.5 mL 加入pH 1.2盐酸溶液100 mL,在(37±0.5) °C下通过磁力搅拌(100 r·min-1)温和混合,观察形成微乳的状态,并在伪三元相图上标出能够形成透明或蓝白色微乳的区域,连接图点以确定能够形成微乳的自乳化区域边界,自乳化区域越大表明自乳化能力越强[10]。结果见图1。
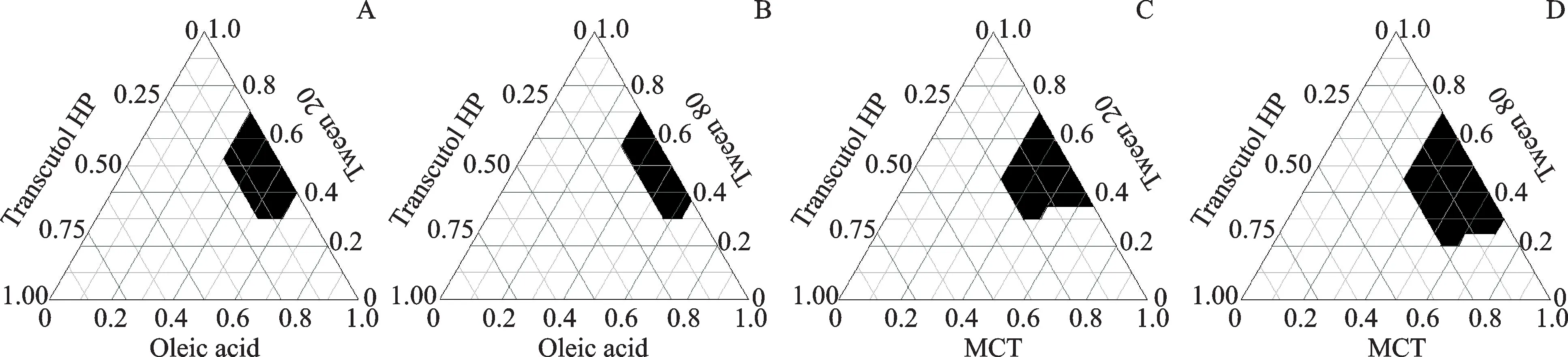
图1 油相、表面活性剂和助表面活性剂构成的伪三元相图Fig.1 Pseudo-ternary phase diagrams composed of oils,surfactant,and co-surfactant
由图1可见,由MCT-Tween 80-Transcutol HP构成的SEDDS形成的自乳化区域面积最大,因此,确定以MCT-Tween 80-transcutol HP 作为CINSEDDS的处方组成,其配比为2∶2∶1。
2.3 CIN-SEDDS的制备
按照处方量精密称取MCT 4 g、Tween 80 4 g、transcutol HP 2 g,置于棕色玻璃瓶中,混合物涡旋5 min,得到透明油状溶液,另精密称取CIN 200 mg加入上述混合物中,搅拌至药物完全溶解,即得CINSEDDS。
2.4 CIN-S-SEDDS的制备
按照2.3项下方法制备CIN-SEDDS,再将沉淀抑制剂加入CIN-SEDDS 中,在室温下涡旋混合5 min,即得CIN-S-SEDDS。
2.4.1 沉淀抑制剂种类筛选 本研究分别以PVP K30、HPMCE5、poloxamer 127和soluplus作为沉淀抑制剂[11]制备CIN-S-SEDDS,沉淀抑制剂加入量均为处方量的5%,通过体外溶出实验确定CIN-SSEDDS处方中沉淀抑制剂的种类。结果见图2。

图2 单一沉淀抑制剂对CIN-S-SEDDS中药物溶出度的影响Fig.2 Effects of single precipitation inhibitor on the dissolution profiles of CIN-S-SEDDS
由图2可见,未加沉淀抑制剂的CIN-SEDDS在pH 1.2盐酸溶液中30 min内药物基本完全溶出,当将溶出介质的pH 调至6.8时,药物溶出度急剧下降至约20%;而以soluplus作为沉淀抑制剂时,在pH 6.8介质溶液中的溶出度明显高于含有其他沉淀抑制剂的CIN-SEDDS。
2.4.2 沉淀抑制剂组合筛选 选择soluplus 与PVP K30、HPMCE5、poloxamer 127 按质量比均为1∶1分别组合作为复合沉淀抑制剂[12]制备CIN-SSEDDS,沉淀抑制剂加入量均为处方质量的5%,并通过体外溶出实验确定CIN-S-SEDDS 处方中沉淀抑制剂的组合。结果见图3。
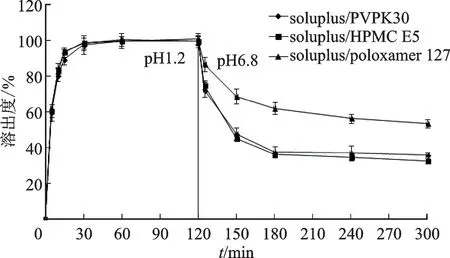
图3 复合沉淀抑制剂对CIN-S-SEDDS中药物溶出度的影响Fig.3 Effects of mixed precipitation inhibitors on the dissolution profiles of CIN-S-SEDDS
由图3可见,以soluplus和poloxamer 127作为复合沉淀抑制剂能够明显抑制药物沉淀。因此,本研究在处方中添加soluplus和poloxamer 127 组合作为复合沉淀抑制剂制备CIN-S-SEDDS。
2.4.3 沉淀抑制剂配比筛选 分别选择soluplus与poloxamer 127按照质量比为1∶3、1∶2、1∶1、2∶1、3∶1形成的组合物作为沉淀抑制剂制备CIN-S-SEDDS,沉淀抑制剂加入量均为处方质量的5%,并通过体外溶出实验确定CIN-S-SEDDS 处方中soluplus 与poloxamer 127的质量比。结果见图4。
由图4可见,soluplus与poloxamer 127的质量比对抑制药物沉淀有很大影响。当soluplus 与poloxamer 127的质量比为2∶1时,CIN-S-SEDDS在pH 6.8介质溶液中的药物溶出度最高。

图4 soluplus与poloxamer 127质量比对CIN-S-SEDDS中药物溶出度的影响Fig.4 Effects of the ratio of soluplus to poloxamer 127 on the dissolution profiles of CIN-S-SEDDS
2.4.4 沉淀抑制剂用量筛选 选择soluplus 与poloxamer 127的质量比为2∶1作为复合沉淀抑制剂制备CIN-S-SEDDS,其用量分别为处方质量的1.0%、2.5%、5.0%、7.5%,通过体外溶出实验确定CIN-S-SEDDS处方中soluplus与poloxamer 127的加入量。结果见图5。
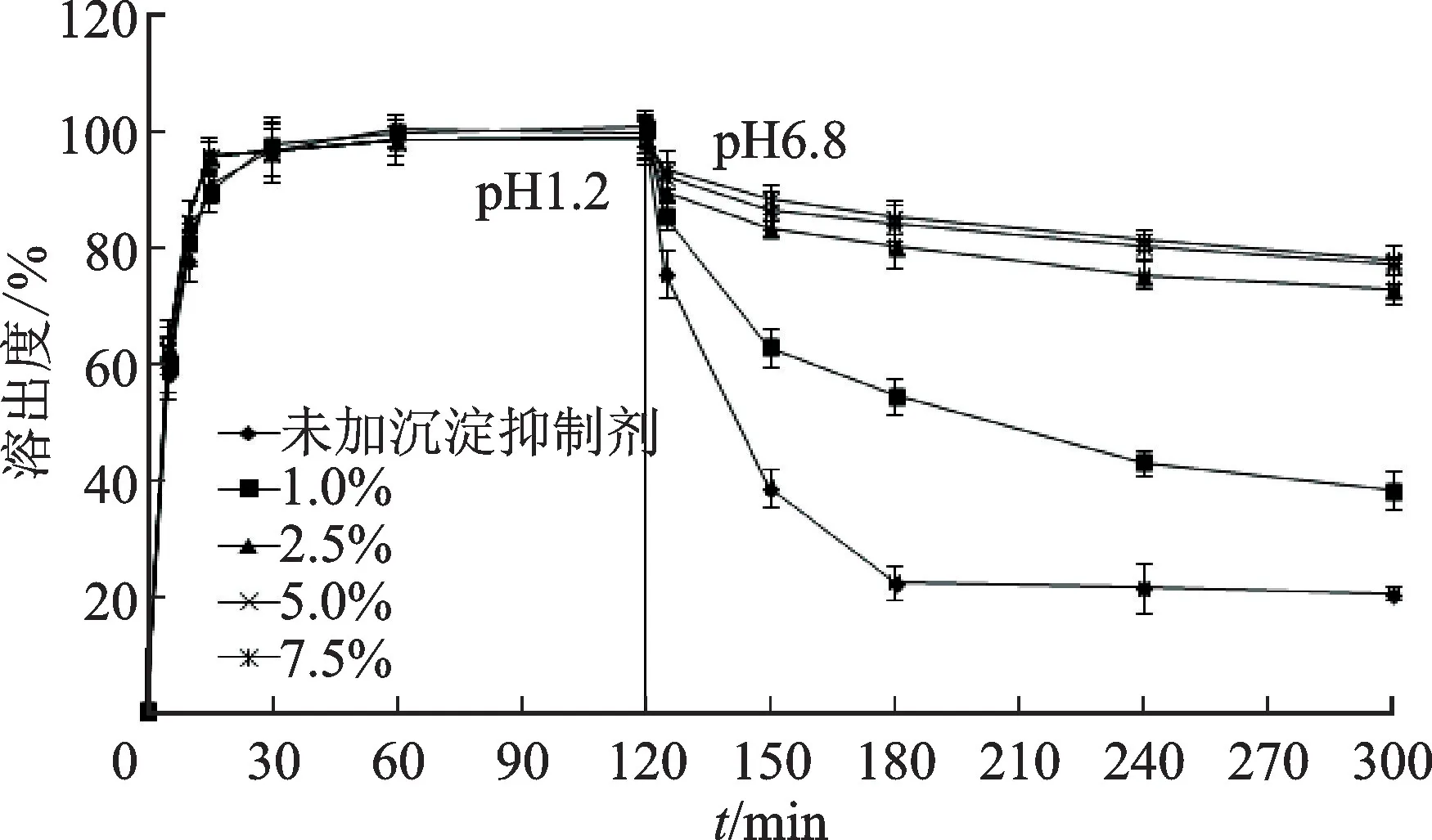
图5 复合沉淀抑制剂用量对CIN-S-SEDDS中药物溶出度的影响Fig.5 Effects of the amount of precipitation inhibitors on the dissolution profiles of CIN-S-SEDDS
由图5可见,处方中soluplus与poloxamer 127的加入量对抑制药物沉淀有很大影响。当soluplus与poloxamer 127 的用量占处方质量比为5%时,CIN-S-SEDDS在pH 6.8溶液中的药物溶出度基本达到最高值。因此,确定soluplus和poloxamer 127的质量比为2∶1、用量占处方质量的5%。
2.5 质量评价
2.5.1 微观形态 用透射电子显微镜观察CINSEDDS、CIN-S-SEDDS 形成的微乳微观形态。用pH 1.2 盐酸溶液分别稀释CIN-SEDDS 和CIN-SSEDDS,使其各自形成淡蓝色微乳,取2份微乳滴加到涂有碳膜的铜网上,用滤纸在样品边缘吸取多余水分,滴加质量浓度为30 mg·mL-1磷钨酸溶液,染色5 min,将样品置于阴凉处晾干,用透射电镜观察2份微乳的微观形态,并拍照,见图6。由图6可见,CINSEDDS和CIN-S-SEDD 经pH 1.2盐酸溶液稀释后均形成直径小于150 nm 的均匀球形微乳,且二者的微观形态几乎无差异,表明沉淀抑制剂的加入对微乳性质的影响较小。

图6 CIN-SEDDS和CIN-S-SEDDS形成微乳的透射电镜照片Fig.6 TEM images of microemulsion formed by CIN-SEDDS and CIN-S-SEDDS
2.5.2 粒径分布、Zeta电位 取CIN-SEDDS和CINS-SEDDS少量,用pH 1.2盐酸溶液分别稀释形成微乳,取2份微乳液体测定粒径分布与Zeta电位。结果显示,CIN-SEDDS、CIN-S-SEDDS经pH 1.2盐酸溶液稀释后,形成的微乳粒径分别为(109.2±4.8)、(104.3±3.9) nm,PDI分别为(0.268±0.009)、(0.271±0.013),Zeta电位分别为(-32.1±0.9)、(-27.1±0.7) mV。
2.6 体外溶出度研究
通过两步溶出度方法比较桂利嗪片、CINSEDDS和CIN-S-SEDDS 的体外溶出情况[13],首先选用pH 1.2盐酸溶液450 mL作为第一步溶出介质(模拟胃液环境),将CIN-S-SEDDS 加入溶出杯中,2 h后再将0.2 mol·L-1磷酸氢二钠溶液450 mL 加入溶出杯中,调节pH 值至6.8(模拟肠液环境),继续进行溶出实验,搅拌桨转速为(50±1) r·min-1,温度为(37±0.5) °C,在规定的时间点(5、10、15、30、60、120、125、150、180、240、300 min)取介质溶液3 mL(补加同温同体积对应空白介质溶液),经0.22 μm 微孔滤膜过滤、稀释后,进样检测药物含量。结果见表2。
由表2 可知,在pH 1.2 盐酸溶液中,CINSEDDS和CIN-S-SEDDS中的药物均可在15 min内完全溶出,而桂利嗪片中药物的溶出较缓慢,在60 min内药物才完全溶出;溶出介质的pH 值升高至6.8时,桂利嗪片中已溶出的药物含量迅速下降,180 min后药物溶出度仅为10.9%,且在溶解杯中能够明显观察到有药物晶体析出。这是由于CIN 的溶解度具有pH 依赖性,在pH 1.2盐酸溶液中药物的溶解度较高,可完全溶解,但是pH 为6.8后,药物的溶解度较低,药物达到过饱和状态,导致药物析出沉淀。在180 min时CIN-SEDDS 中药物的溶出度降低到22.1%,CIN-S-SEDDS中药物的溶出度降低到84.1%,CIN-S-SEDDS中已溶出的药物含量明显高于CIN-SEDDS和桂利嗪片,表明CIN-S-SEDDS 中的沉淀抑制剂可稳定药物,使其始终处于过饱和状态,抑制药物聚集成沉淀,这些处于溶解状态的药物在进入肠液后可被充分吸收和利用,进而提高药物的口服生物利用度。
表2 桂利嗪片、CIN-SEDDS和CIN-S-SEDDS在模拟体内介质中的药物溶出度 (n=6,)Tab.2 Dissolution of CIN-SEDDS,CIN-S-SEDDS,Cinnarizine Tablets in simulated in vivo media (n=6,)

表2 桂利嗪片、CIN-SEDDS和CIN-S-SEDDS在模拟体内介质中的药物溶出度 (n=6,)Tab.2 Dissolution of CIN-SEDDS,CIN-S-SEDDS,Cinnarizine Tablets in simulated in vivo media (n=6,)
3 讨论
SEDDS是由油相、表面活性剂、助表面活性剂和药物组成的各相同性混合物,加水稀释后在温和搅拌下即可形成纳米级微乳,SEDDS已成为改善难溶性药物口服生物利用度最有效手段之一[14]。然而,某些药物通过SEDDS口服给药后,容易析出沉淀,不利于药物吸收,通过在SEDDS 的处方中添加PVP K30、HPMC、CMC-Na、soluplus、泊洛沙姆等聚合物,可使难溶性药物处于并保持过饱和状态,延缓或阻碍药物析出沉淀,以增加药物的吸收量,这类聚合物被称为沉淀抑制剂[15]。沉淀抑制剂主要通过热力学和动力学2种机制延缓或阻碍药物析出沉淀[16]。热力学过程主要是提高药物的溶解度,进而降低过饱和度[17]。动力学过程主要是通过聚合物与药物分子之间的氢键、疏水作用力、空间位阻、增加溶液黏度等方式抑制药物聚集和沉淀[18]。
本文采用系统的处方开发方法将桂利嗪制备成过饱和自乳化释药系统,虽然该处方能够有效降低药物在pH 6.8介质溶液中的沉淀析出量,但是CIN-SSEDDS为液体状态,其在储存过程中存在一定的不稳定性,需要将其制备成固体制剂以便提高制剂的稳定性,方便携带、服用。因此,后续将对CIN-SSEDDS的固体化工艺进行研究。
猜你喜欢
皮革制作与环保科技(2022年18期)2022-11-26
新材料产业(2022年2期)2022-07-19
当代化工(2019年4期)2019-12-03
商情(2019年43期)2019-10-20
智富时代(2018年6期)2018-08-06
智富时代(2018年6期)2018-08-06
试题与研究·中考化学(2016年4期)2017-03-28
中学化学(2016年10期)2017-01-07
中学生数理化·中考版(2015年11期)2015-09-10
中学化学(2015年5期)2015-07-13
