
药用辅料级枸橼酸三乙酯工艺研究
2022-08-02 05:13:46赵明王雪志缪志毅
药品评价 2022年9期
赵明,王雪志,缪志毅
1.江西省药品检查员中心第四检查所,江西 南昌 330000;2.江西阿尔法高科药业,江西 萍乡 337000
枸橼酸三乙酯又称柠檬酸三乙酯,为一种常见的增塑剂,可增加药物制剂塑性,涉及胶囊剂、片剂等剂型,如制备阿司匹林肠溶片等[1]。枸橼酸三乙酯在食品行业也作添加剂,可与水、乙醇等组成的无表面活性剂形成微乳液[2]。在日化工业中可和三乙酸甘油酯作为增塑剂配合使用[3]。因其广泛的用途,有关其合成方法,深受学者们关注。
工业级的枸橼酸三乙酯的合成方法很多。在较早的时候,刘欣宇等[4]在浓硫酸、磷酸、苯磺酸等混酸作用下,以甲苯为分水剂制备高纯度枸橼酸三乙酯,此工艺存在甲苯残留等问题,且后处理经过萃取、蒸馏等繁琐步骤,工艺复杂。后来更多作者报道[5-7]采用固体酸催化剂催化制备得到柠檬酸三乙酯。然而反应温度均需要在120~130 ℃下进行,因乙醇体系的沸点较低,很难达到此温度范围,不可避免地引入了分水剂,以便达到升温分水的效果,此类工艺能耗高且后处理复杂,彭文勇等[8]利用对甲苯磺酸和固体酸混合催化制备得到的柠檬酸三乙酯,同样存在以上问题。黄飞等[9]采用微波催化的方法合成了柠檬酸三乙酯,但该方法能耗高且同样需要在酸性催化下进行,该法只是促进了酯化的进行,并未解决如上问题,另外药用级枸橼酸三乙酯在各国药典中质量要求相当严格,均要求其最大单杂不得大于0.2%,而市售工业级产品很难达到其纯度要求。所以寻找合适的符合药用辅料级产品的制备工艺,已成近年来的焦点。本研究开发了药用辅料枸橼酸三乙酯的制备工艺,工艺简单,收率高,产品质量高,可实现连续化大生产,是一种优良的大生产工艺。
1 材料与方法
1.1 主要原材料
枸橼酸(南京化学试剂股份有限公司,分析纯,99.9%);二氯甲烷(天津富宇精细化工,分析纯,99.8%);无水乙醇(南京化学试剂股份有限公司,分析纯,99.5%);对甲苯磺酸(上海麦克林试剂生物科技有限公司,分析纯,98%);活性白土(分析级);亚磷酸(上海阿拉丁生化科技股份有限公司,分析纯,99%);枸橼酸三乙酯对照品(中国食品药品检定研究院,纯度99.6%)等。
1.2 主要实验仪器
10 L 三口反应釜、球形冷凝管、温度计、层析过滤柱、脱水膜管;气相色谱仪[型号:岛津(中国)有限公司,GC-2018];红外光谱仪[型号:布鲁克(北京)科技有限公司,TENSOR 27]等。
1.3 制备机制
本研究采用对甲苯磺酸和亚磷酸为催化剂,枸橼酸在过量的乙醇中回流反应,通过膜脱水技术控制反应体系水分,制备得到枸橼酸三乙酯。反应方程式见图1。
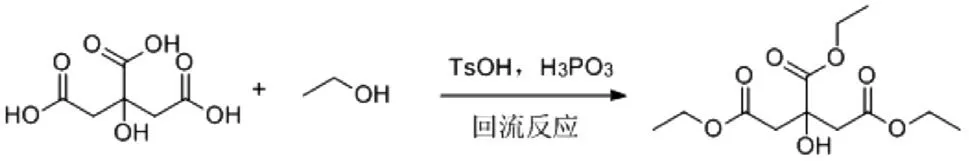
图1 枸橼酸三乙酯反应方程式
1.4 工艺方法
1.4.1 枸橼酸三乙酯制备工艺将一定量的枸橼酸、乙醇加入反应瓶中,加入一定量的对甲苯磺酸和亚磷酸,搅拌升温至整个体系出现强烈回流为止,回流的乙醇通过膜脱水处理,泵回反应釜,保温反应一定时间,中控反应液酸值,酸值合格后,反应液降温至室温,搅拌下缓慢滴入和对甲苯磺酸等当量的30%氢氧化钠水溶液,室温搅拌约30 min,反应液再通过减压浓缩脱去溶媒后,并控温80℃减压进一步脱去低沸物,反应液通过特定精制柱,过滤,得高纯度枸橼酸三乙酯。
1.4.2 酸值测定参照《中国药典》2020 年版四部[10](通则0713)方法检测。
1.4.3 有关物质测定(最大单杂、总杂)取本品,加二氯甲烷溶解并稀释制成每1 mL 含30 mg 的溶液,作为供试品溶液;精密量取1 mL,置100 mL量瓶中,加二氯甲烷稀释至刻度,摇匀,作为对照溶液。照含量测定项下的色谱条件测定。
1.4.4 含量测定取本品约300 mg,精密称定,加二氯甲烷溶解并稀释制成每1 mL 中含30 mg 的溶液,精密量取1 μL 注入气相色谱仪,记录色谱图;另取枸橼酸三乙酯对照品适量,同法测定,按外标法以峰面积计算,即得。
1.4.5 收率的计算

公式中276.29 为枸橼酸三乙酯分子量;192.13为枸橼酸分子量。
2 结果与讨论
2.1 单因素实验
2.1.1 原料配比优化实验将不同质量比的枸橼酸、乙醇加入反应瓶中,暂选择亚磷酸和对甲苯磺酸质量比为1∶3 作为催化剂,催化剂总质量为枸橼酸质量的3%,升温至回流,回流的乙醇通过膜脱水处理控制水分,保温反应10 h,最终反应液脱溶除水后,再通过同等精制柱进行精制处理,得产品枸橼酸三乙酯,以最终产品最大单杂、收率、产品色泽和含量为对比指标,通过综合分析,考察不同原料配比对反应结果的影响,结果见表1。
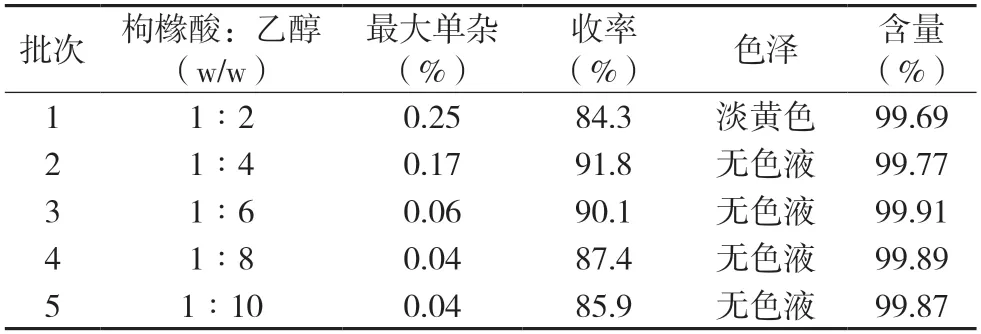
表1 物料配比对反应结果的影响
从表1 中可以看出,乙醇量较少的情况下,随着反应的进行,整个体系含水率偏高,抑制了酯化的进一步进行,造成收率低;也因为整个体系酸浓度高,副反应增多,最终产品带有少许色泽。反而随着溶剂的增大,酸浓度下降,酯化率下降,造成收率低。枸橼酸和乙醇质量比1∶4 的情况下,虽然产品收率最高,但是有关杂质接近药典的限度要求≤0.2%,故最优的物料配比为橼酸和乙醇质量比为1∶6。
2.1.2 酯化时间优化实验在常见的酯化反应中,酯化时间越长收率越高,但副产物也会越多,酯化的时间对收率和杂质影响最大,以最终产品最大单杂、收率、产品色泽和含量为对比指标,我们选择不同的酯化时间,按照枸橼酸、乙醇质量比1∶6的比例加入反应瓶中,亚磷酸和对甲苯磺酸质量比为1∶3 作为催化剂,催化剂总质量为枸橼酸质量的3%,最终反应液脱溶除水后,再通过同等精制柱进行精制处理,得产品枸橼酸三乙酯,考察酯化时间对反应影响,结果见表2。由表2 可见,随着酯化时间加长,产品颜色基本无区别,这归功于反应过程大量乙醇和亚磷酸的保护,并经精制柱的处理后产品颜色差异不大。但是随着反应时间的变化,收率和产品纯度也随之变化,由上表可知,最佳的酯化时间为12 h。

表2 酯化时间对反应结果的影响
2.1.3 催化剂的选择优化实验酯化反应往往最关键的就是催化剂的选择,但药用辅料级产品和工业级产品的工艺重心有所不同,工业级产品往往只关注产品收率和产品的纯度指标,而药用辅料级产品除了收率和产品纯度,更关注的还是产品的安全性,通常要牺牲收率以便最终产品更加安全。对催化剂的选择进行充分研究,以产品最终的收率,最大单杂为对比指标,按照枸橼酸、乙醇质量比1∶6 的比例加入反应瓶中,加入不同的催化剂,升温至回流,回流的乙醇通过膜脱水处理控制水分,保温反应12 h,最终反应液脱溶除水后,再通过同等精制柱进行精制处理,得产品枸橼酸三乙酯;采用《中国药典》2020 年版四部枸橼酸三乙酯的有关物质检测方法[10]进行分析,结果见表3。

表3 催化剂对反应结果的影响(%)
由表3 可见,亚磷酸可作为保护剂和催化剂参与酯化反应,选择对甲苯磺酸+亚磷酸的混合催化剂效果最好;当甲苯磺酸和亚磷酸的质量比为9∶1,催化剂用量为枸橼酸的质量比3%时,产品最大单杂和最终收率均较理想,同时产品经纯化工序,产品中杂质基本被精制去除,安全性高。
2.2 后处理精制优化实验
2.2.1 精制柱的制备取层析桶,底部加滤纸,滤纸上层垫好一层脱脂棉,加入枸橼酸投料量的约1 倍量的颗粒状活性炭,然后上层再垫一层脱脂棉,把小一号的底部带孔可提滤桶加在活性炭层上层,滤桶同样加上滤纸,滤纸上层加上枸橼酸投料量的约1 倍量的氧化铝粉末,上层垫一层脱脂棉,压实,制成的精制柱备用。
2.2.2 后处理优化实验按照上述优化后的最优条件,按枸橼酸、乙醇质量比1∶6 的比例加入反应瓶中,加入总用量为枸橼酸的质量比3%的甲苯磺酸、亚磷酸混合催化剂,回流保温反应12 h,最终反应液脱溶除水后得粗品。粗品等量分成三份:一份未通过精制柱过滤;一份经过精制柱进行处理;另一份通过精馏纯化,制得产品;分别计算收率,同时最终的三份产品进行质量对比研究,结果见表4。

表4 精制方法对产品质量的影响
从表4 可知,经特定的精制柱,最终产品质量得到有效提升,产品的酸值和单杂降低效果明显,主要的原因是经精制柱可有效地去除副产物枸橼酸二乙酯和枸橼酸单乙酯等杂质,另采用精馏方法处理可得到更高质量的注射级枸橼酸三乙酯。最终,采用精制柱纯化所得到的产品经全检,其质量符合药用辅料的技术要求。
2.3 产品分析
对采用精制柱纯化所得药用辅料级枸橼酸三乙酯进行红外解析,见图2。

图2 精制柱纯化的枸橼酸三乙酯红外色谱图
(1)3 497 cm-1:-O-H 伸缩振动;1 342 cm-1:O-H弯曲振动;1 115 cm-1:C-O 伸缩振动,证明结构中含叔 醇(-OH)结 构;(2)2 985、2 941、2 909、2 877 cm-1:C-H 伸缩振动;1 468、1 447、1 372 cm-1:C-H弯曲振动,证明本品结构中含亚甲基和甲基结构;(3)1 738 cm-1:C=O 伸缩振动;1 196、1 098 cm-1:C-O-C 不对称伸缩振动,证明本品结构中含酯基(RCOOR’)结构。由红外光谱可知,本品结构中含叔醇(-OH)、酯基(RCOOR’)、亚甲基和甲基结构等。上述红外光谱数据与枸橼酸三乙酯结构相符。
3 结论
对药用辅料级枸橼酸三乙酯进行详细的技术开发研究,最终得出最佳的制备工艺,选择原料枸橼酸和乙醇质量比1∶6,以甲苯磺酸、亚磷酸质量比9∶1 为混合催化剂,加入总用量为枸橼酸的质量比3%,升温至回流,回流的乙醇通过膜脱水处理控制水分,保温反应12 h,反应液经过初步中和、脱溶、除水后得粗品,粗品再通过特定的精制柱可制得药用辅料级枸橼酸三乙酯;该制备工艺简单,收率高,产品质量高,可实现连续化大生产,是一种优良的大生产工艺。
猜你喜欢
肝博士(2024年1期)2024-03-12 08:39:04
基层中医药(2022年7期)2022-11-17 08:25:08
无机盐工业(2020年6期)2020-06-12 03:35:16
山东化工(2020年4期)2020-03-30 08:39:38
医药前沿(2018年2期)2018-01-17 12:10:56
中成药(2017年10期)2017-11-16 00:50:02
实用临床医学(2016年8期)2016-06-07 01:28:16
合成技术及应用(2015年3期)2015-12-11 08:36:28
中国塑料(2015年8期)2015-10-14 01:10:44
河南科技(2014年19期)2014-02-27 14:15:32
