
7-硝基苯并噻吩-2-甲酸乙酯合成实验的优化与改进
2022-08-01 06:56:56李家柱洪莹莹满英秀李英豪李庆忠何涛
大学化学 2022年5期
李家柱,洪莹莹,满英秀,李英豪,李庆忠,何涛
烟台大学化学化工学院,山东 烟台 264005
1 引言
有机合成实验是衔接化学专业本科生基础实验和毕业论文的重要环节,旨在通过实验教学,使学生训练并掌握有机合成的基本技能,学会正确选择有机化合物的合成、分离、提纯和分析鉴定的方法。基础有机实验主要针对学生的基本实验技能和单元反应操作进行训练,而有机合成实验在此基础上更注重综合研究能力的培养。多步有机合成反应环环相扣,如果某步反应没有产物或产物纯度不够,会导致后续合成的失败,因此多步有机合成有利于培养学生良好的实验习惯及严谨求实的科学作风,也有利于提高学生分析问题、解决问题的综合实验能力[1,2]。
苯并噻吩及其衍生物在自然界中广泛存在,是含硫原子的芳香杂环化合物之一。苯并噻吩衍生物在工业中的用途广泛,可以作为染料、药物及有机发光半导体材料等[3]。很多天然产物和药物分子含有苯并噻吩结构,如齐留通(Zileuton)具有抗哮喘作用[4],2014年上市的新药伊格列净(Ipragliflozin)可用于治疗II型糖尿病[5]。文献报道的苯并噻吩制备方法很多,区别在于原料选择的不同,如从苯的衍生物合成,由噻吩衍生物合成以及由其他杂环合成等[3]。
7-硝基苯并噻吩-2-甲酸乙酯的合成是本校应用化学专业学生有机合成实验中的综合训练项目。原合成路线如图1所示,该路线存在耗时较长、产率不稳定,后处理难度较大及使用有毒试剂等问题。本项目旨在改进苯并噻吩环系的合成方法和步骤,降低试剂用量,提高产率,优化后处理过程,减少有毒物使用和三废排放,使之更符合教学实验和绿色化学要求;同时,改进后的实验增加了有机波谱分析内容,训练学生通过红外光谱、核磁共振氢谱及质谱等方式对目标化合物进行表征,更有利于学生综合素质的培养。
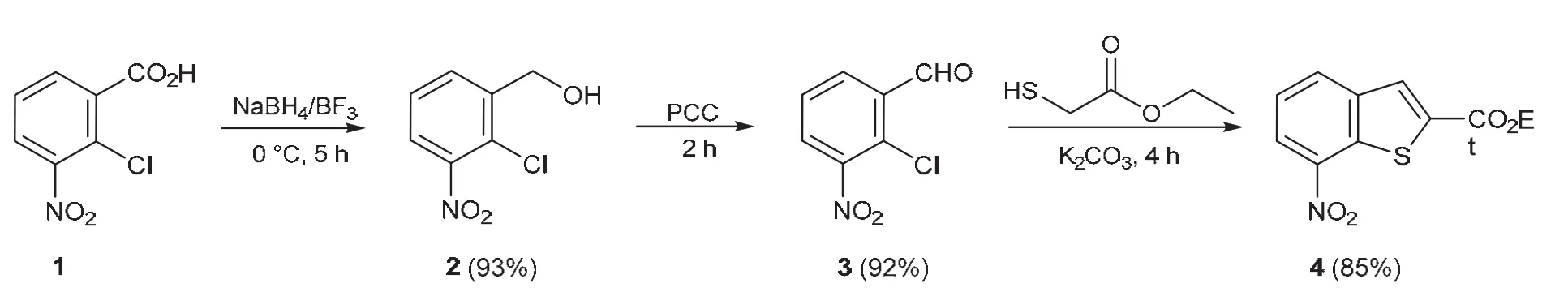
图1 改进前7-硝基苯并噻吩-2-甲酸乙酯的合成路线
2 实验部分
2.1 实验原理
本实验采用水杨醛为起始原料,通过硝化、磺酰化、芳基亲核取代和脑文格反应的方法来制备7-硝基苯并噻吩-2-甲酸乙酯。改进后合成路线如图2所示,水杨醛(5)经硝化反应得到3-硝基水杨醛(6)[6],在碱性条件下与对甲苯磺酰氯(TsCl)反应后得磺酰化产物(7),最后在弱碱碳酸钾存在条件下与巯基乙酸乙酯反应即得目标化合物7-硝基苯并噻吩-2-甲酸乙酯(4)。
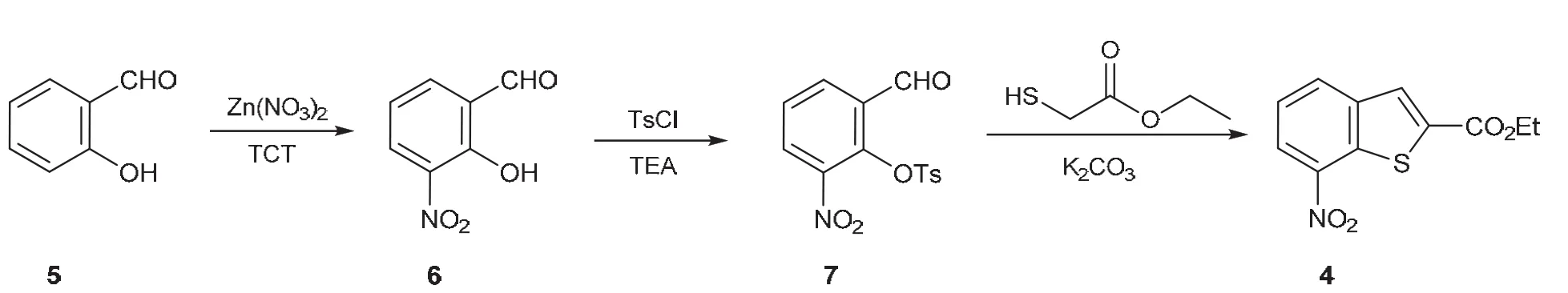
图2 改进后7-硝基苯并噻吩-2-甲酸乙酯的合成路线
关键步骤苯并噻吩环的合成机理如图3所示,反应底物3或7的离去基团两个邻位分别被吸电子基团醛基和硝基取代,可被巯基乙酸乙酯在碳酸钾作用下生成的亲核性硫负离子进攻,从而发生芳基亲核取代反应生成中间体A,酯基α-位亚甲基同时与吸电子的硫原子相连,α-H酸性较强,碳酸钾拔氢后得到碳负离子B,亲核进攻羰基碳后得到中间体C,最后经质子转移(得D)、脱水两步得到目标化合物4。
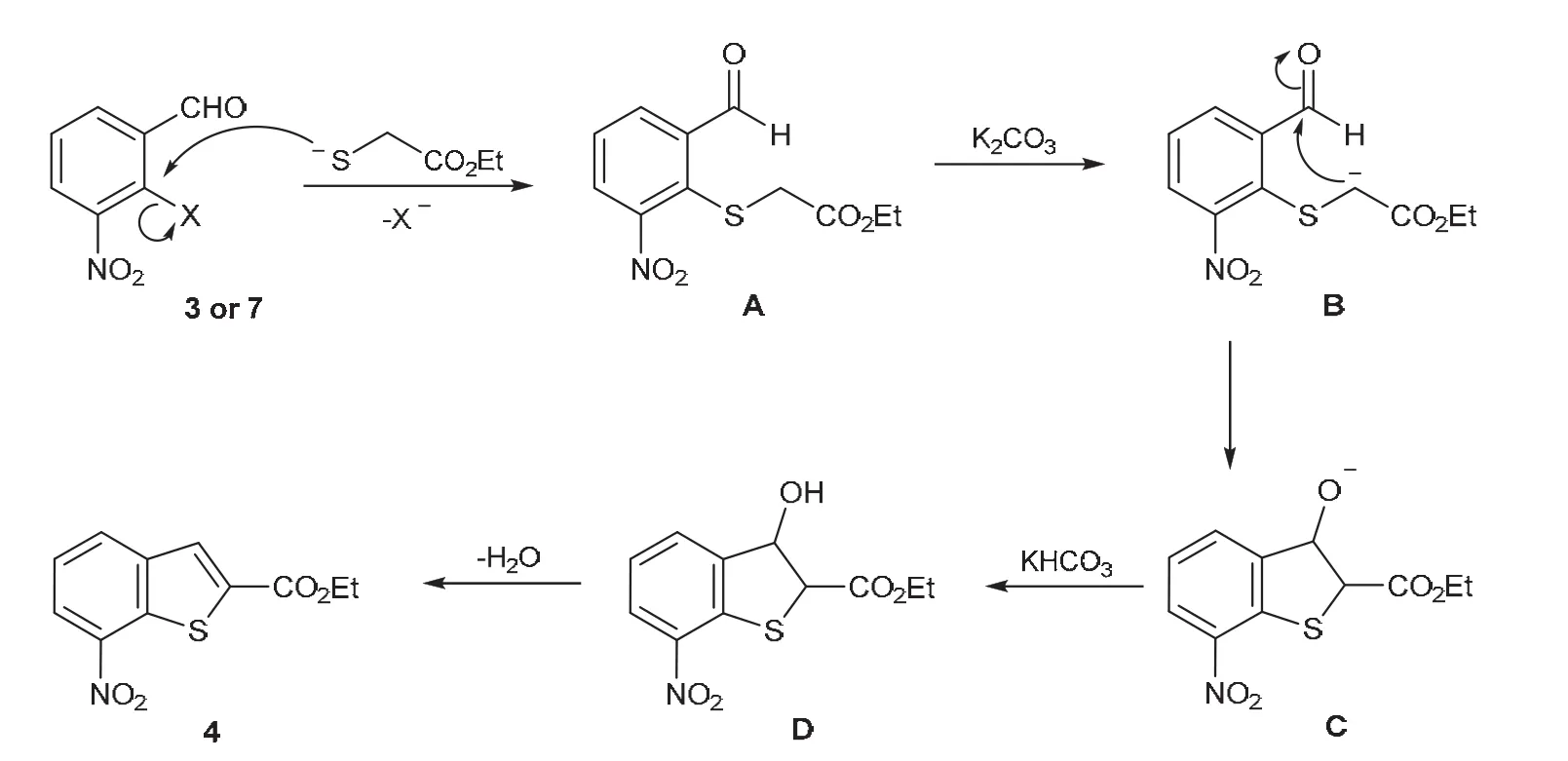
图3 7-硝基苯并噻吩-2-甲酸乙酯合成反应机理
2.2 试剂和材料
水杨醛、硝酸、对甲苯磺酰氯、巯基乙酸乙酯、四氢呋喃、乙腈、丙酮、乙酸乙酯、碳酸钾、硝酸锌、三氯三嗪、饱和Na2CO3、无水硫酸钠、二氯甲烷,均为国药分析纯。GF254高效薄层层析硅胶板,柱层析硅胶(100–200目),均购自江友硅胶制品厂。
2.3 仪器和表征方法
圆底烧瓶,翻口塞、注射器、锥形瓶、玻璃棒、磁子、布氏漏斗、抽滤瓶、磁力搅拌器、恒温磁力搅拌油浴锅、循环水式真空泵、旋转蒸发仪,WRS-2A型显微熔点测定仪,Nicolet Nexus-670傅立叶红外光谱仪,Bruker 400-MR型核磁共振仪,AB SCIEX API 4000液质联用仪。
2.4 实验步骤和方法
2.4.1 3-硝基水杨醛(6)的合成
量取水杨醛(1.22 g,10 mmol)加入到圆底烧瓶中,放入磁子,搅拌加入乙腈(50 mL)使原料溶解,然后加入六水硝酸锌(6.06 g,20 mmol)和三聚氯氰(TCT,303 mg,1.6 mmol),在50 °C下反应1 h,TLC检测原料消失后结束反应。减压浓缩,残余物用乙酸乙酯溶解后加水萃取,合并有机相,无水硫酸钠干燥,旋干后干法上样,通过柱层析分离(洗脱剂:V(石油醚) :V(乙酸乙酯) = 20 : 1)得到黄色固体1.35 g (81%)。
2.4.2 化合物7的合成
将3-硝基水杨醛(1.0 g,5.98 mmol)溶于二氯甲烷(20 mL),体系为黄色溶液,加入三乙胺(1.5 mL),体系颜色变橘黄色,将体系移至冰水浴,控制温度10 °C以下,分批加入对甲苯磺酰氯(TsCl,1.20 g,6.28 mmol),反应10 min后取样点板,反应结束。将体系用1 mol·L-1稀HCl调pH < 7,乙酸乙酯萃取,有机相用无水硫酸钠干燥,抽滤后滤液减压浓缩,用乙醇重结晶后得黄色固体产物1.88 g (98%),直接用于下一步反应。
2.4.3 7-硝基苯并噻吩-2-甲酸乙酯(4)的合成
将上一步产物7 (1.8 g,5.60 mmol)溶于THF (20 mL),体系为黄色溶液,依次加入K2CO3(2.69 g,22.41 mmol)与巯基乙酸乙酯(1.5 mL),升温回流(体系颜色由淡黄色逐渐变为橙红色),3 h后TLC点板,反应结束。加水和乙酸乙酯分出有机相,水洗,有机相用无水硫酸钠干燥,减压浓缩,用DCM/石油醚沉析,得黄色固体1.34 g (95%)。mp 189.1–190.0 °C。1H NMR (400 MHz, CDCl3)δ8.49 (dd,J= 8.0, 0.8 Hz, 1H, 6-H),8.20 (dd,J= 7.9, 1.0 Hz, 1H, 4-H),8.15 (s, 1H, 3-H),7.59 (t,J= 7.9 Hz, 1H,5-H),4.44 (q,J= 7.1 Hz, 2H, OCH2CH3),1.44 (t,J= 7.1 Hz, 3H, OCH2CH3)。MS (ESI): calcd.for C11H9NO4S 251.0;foundm/z252.1 (MH+)。
3 结果与讨论
3.1 改进前合成路线
改进前合成路线如图1所示,以2-氯-3-硝基苯甲酸(1)为初始原料,经NaBH4/BF3·OEt2还原后得2-氯-3-硝基苄醇(2),氯铬酸吡啶鎓盐(PCC)氧化后得2-氯-3-硝基苯甲醛(3),最后与巯基乙酸乙酯反应后得到目标产物7-硝基苯并噻吩-2-甲酸乙酯(4)。该路线主要存在以下问题:(1) 反应时间较长,如第一步反应需要5 h完成;(2) 用到毒性较大的PCC,且反应后产物吸附严重,分离较困难,需要大量二氯甲烷冲洗;(3) 最后一步反应合成目标产物时所用溶剂为高沸点的DMF,后处理过程需要多次萃取,耗时费力。学生完成整个实验流程需要24学时。
3.2 改进思路
通过对目标产物苯并噻吩合成反应机理的分析(图3),发现其关键步骤在于巯基乙酸乙酯与前体的芳基亲核取代(SNAr)反应,其中离去基团X若为Cl,则通过逆合成分析可推出初始原料为原路线所用2-氯-3-硝基苯甲酸(1)。根据有机化学基础理论,离去基团的离去倾向与其离去后形成的负离子碱性有关。一般来说,离去基团离去后所形成负离子的碱性越弱,其离去能力越强。除了卤素之外,磺酸酯基也是常见离去基团,因此我们设想,如果将离去基团X由氯换成对甲苯磺酰氧基(TsO―),也应可发生芳基亲核取代反应,从而用于苯并噻吩环系的合成。而X换为TsO―后可通过逆合成分析顺利推导出常见原料水杨醛(图4)。
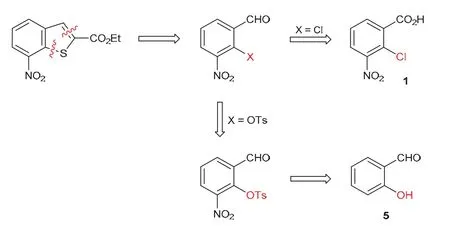
图4 7-硝基苯并噻吩-2-甲酸乙酯的逆合成分析
3.3 改进后合成路线
改进后合成路线如图2所示,以水杨醛(5)为原料,经六水合硝酸锌选择性硝化得到3-硝基水杨醛(6),在三乙胺存在条件下与对甲苯磺酰氯(TsCl)反应后得磺酰化产物(7),最后在弱碱碳酸钾存在条件下与巯基乙酸乙酯连续发生芳基亲核取代反应和脑文格反应即得目标化合物7-硝基苯并噻吩-2-甲酸乙酯(4),学生完成整个实验流程仅需要12学时。
目标产物4用乙醇重结晶后测得熔点为189.1–190.0 °C,与改进前产物数据一致。核磁共振氢谱如图5所示,在7.5–8.5之间出现四组峰,归属为7-硝基苯并噻吩-2-甲酸乙酯芳环上4个氢质子吸收,根据偶合裂分关系,其中8.15处的单峰(1H)归属为噻吩环上3-H;7.59处出现的三重峰(1H)归属为苯环上的5-H,因其被4-H和6-H裂分;8.49和8.20处出现的两组双重峰(积分均为1H)可分别归属为6-H和4-H,其中6-H受硝基吸电子诱导作用影响较大,出现在最低场(8.49),这三组峰的偶合常数J均在7.9–8.0 Hz范围内,典型的苯环邻位偶合关系。而在高场区,4.44的四重峰(2H)和1.44处的三重峰(3H)相互偶合(J= 7.1 Hz),分别归属为乙酯结构中的CH2和CH3。目标产物4的红外光谱如图6所示,酯羰基伸缩振动出现在1700.9 cm-1,硝基伸缩振动出现在1517.9和1322.9 cm-1,而苯环上C―H伸缩振动出现在3104.9 cm-1。目标产物4的质谱如图7所示,m/z252.1是该化合物的质子化分子离子峰(MH+)。核磁共振氢谱、红外光谱及质谱特征均与目标产物相符,验证了改进方法的正确性。
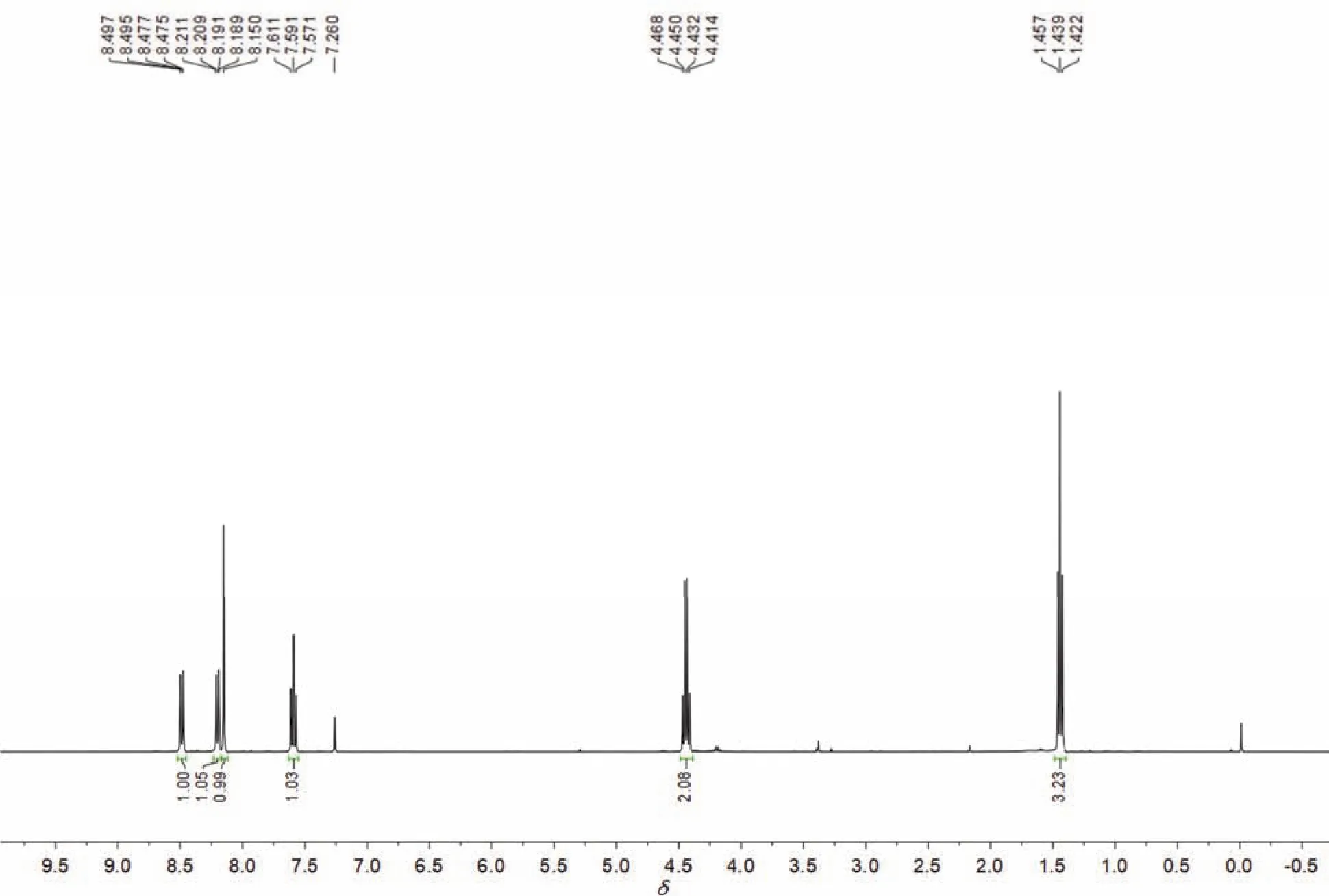
图5 7-硝基苯并噻吩-2-甲酸乙酯的核磁共振氢谱
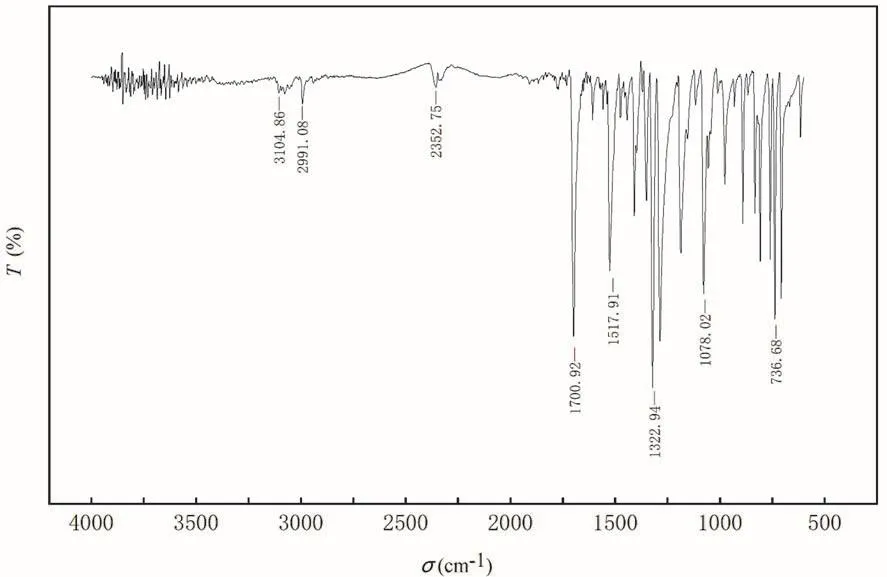
图6 7-硝基苯并噻吩-2-甲酸乙酯的红外光谱
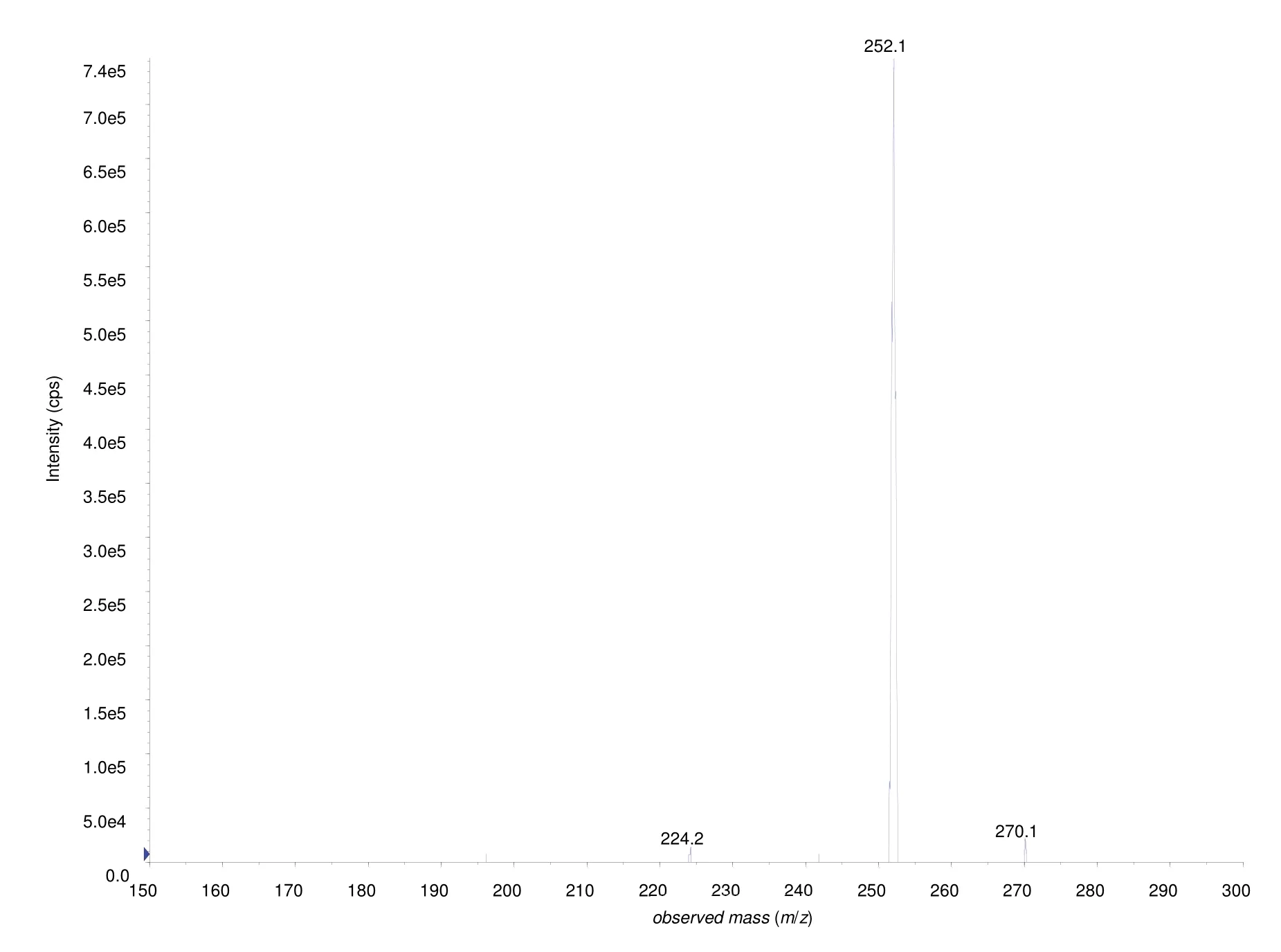
图7 7-硝基苯并噻吩-2-甲酸乙酯的质谱
合成路线改进前后对比如表1所示,改进后的实验方案避免了高毒试剂PCC和高沸点溶剂DMF的使用;整个合成过程无产物吸附现象,后处理过程简单,更符合绿色化学的发展趋势;而且改进后节省了时间,学生在12学时内即可完成整个流程,更适合教学实验的要求。

表1 合成路线改进前后对比
4 结语
针对7-硝基苯并噻吩-2-甲酸乙酯的合成原实验方案的缺陷,本项目首先针对关键步骤苯并噻吩环系的合成进行了分析和改进,以对甲苯磺酰氧基(TsO―)代替氯原子作为离去基团,低沸点的四氢呋喃代替高沸点的DMF作为溶剂完成苯并噻吩合成,以此为基础进行逆合成分析,成功完成以常见化合物水杨醛为起始原料,三步合成目标产物的方法。改进后的合成路线避免了高毒试剂PCC,高沸点溶剂DMF的使用,降低了试剂用量,优化了后处理过程,减少了有毒物使用和三废排放,更符合教学实验和绿色化学要求。同时,改进后的实验通过熔点、红外光谱、核磁共振氢谱及质谱等方式对目标化合物进行表征,涵盖了有机化合物合成的整个研究过程,更有利于学生综合素质的培养。
猜你喜欢
工业用水与废水(2021年4期)2021-09-07 08:56:42
绥化学院学报(2017年9期)2017-09-22 04:54:25
当代化工研究(2016年1期)2016-03-16 22:00:24
合成化学(2015年10期)2016-01-17 08:56:47
应用化工(2014年1期)2014-08-16 13:34:08
应用化工(2014年4期)2014-08-16 13:23:09
应用化工(2014年10期)2014-08-16 13:11:29
应用化工(2014年9期)2014-08-10 14:05:08
无机化学学报(2014年8期)2014-02-28 17:32:40
中国氯碱(2014年10期)2014-02-28 01:04:59
